الجلاكتوزيمية (بالانجليزية: Galactosaemia، باليونانية:γαλακτόζη + αίμα "جالاكتوز + دم"، تراكم الجالاكتوز بالدم) هو اختلال وراثي نادر ينتج عنه خلل بقدرة الجسم على هضم الجالاكتوز ويؤثر على عملية تأييض سكر الجالاكتوز بحيث يمنع حدوثها بطريقة صحيحة، هذا الاختلال الوراثي ينتج عن صفة متنحية تتسبب في نقص بالإنزيم المسؤول عن التحلل الكافي للجالاكتوز.
| الجلاكتوزيمية | |
|---|---|
 | |
| معلومات عامة | |
| الاختصاص | علم الغدد الصم |
| المظهر السريري | |
| الأعراض | عدم تحمل اللاكتوز |
يعتبر الطبيب الألماني فريديش جوبرت Friedrich Goppert (1870- 1927)، أول شخص قام بتشخيص هذا المرض سنة 1917،[1] أما السبب( خلل في تأييض الجالاكتوز) فتم تحديده من قبل مجموعة كان يترأسها هيرمان كالكار Herman Kalckar في عام 1956.[2]. احتمال الإصابة بهذا الخلل تقريبا 1 لكل 60,000 نسمة بالنسبة للسكان الأوروبيين، أما فيما يخص مناطق سكنية أخرى، معدل الإصابة يختلف، على سبيل الذكر، الجالاكتوسيميا شائعة بمعدل أكبر بمئة مرة (1:480 مولود). [3] بين السكان الايرلنديون (Irish travellers )[4]
السبب
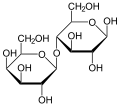
لاكتوز
جلوكوز

جالاكتوز
اللاكتوز في الغذاء (مثل الألبان ومنتجاتها)) يتم تحطيمه إلى مكوناته البسيطة الجلوكوز والجلاكتوز عن طريق انزيم يسمى لاكتيز( Lactase).
في حال الأشخاص الذين يعانون من وجود الجالاكتوز بالدم فإن الإنزيمات اللازمة للقيام بعملية تأييض للجالاكتوز (جالاكتوز -1- فوسفات يوريديل الترانسفيراز) يكون بها نقص بشكل كبير أو مفقود بالكامل مما يؤدي إلى تراكم كمية عالية السمية من (جالاكتوز-1-فوسفات) في الأنسجة، وهذا ما يعرف بال (جالاكتوسوميا الكلاسيكية،)، التي ينتج عنها تضخم الكبد، تليف الكبد، الفشل الكلوي، إعتام عدسة العين، قيء, الاستيلاء, نقص السكر في الدم، الخمول، تلف المخ، وفشل المبيض. بدون علاج، معدل الوفاة بين الاطفال الرضع يصل إلى 75 %.
وجود الجالاكتوز بالدم مرض متعلق بالوراثة والجينات، بحيث أنه يورث من خلال صفة متنحية، مما يعني أن الطفل يجب أن يرث جين مصاب بالخلل من كلا الأبوين حتى يظهر المرض. الأشخاص الذين يكونون مختلفين الزيغوت يصنفون على أنهم حاملون للمرض، لأنهم ورثوا جين طبيعي والجين الاخر كان مصاب، [5] الحاملون للمرض لا تظهر لديهم أية أعراض للمرض .
غياب جين ينتج الانزيم المسؤول عن تحليل الجلاكتوز، والخلل يكون في إحدى المراحل الموجودة ضمن نطاق الدائرة الحمراء في الصورة.

تراكم الجالاكتوز
في مرضى الجلاكتوزيميا، يؤدي تراكم الجلاكتوز إلى تحوله لماده متفاعلة يستهلكها انزيمات في حلقة بوليول (polyol pathway). الخطوة الأولى في هذا المسار هو اختزال الالديهيدات (نوع من أنواع السكر تتضمن الجلاكتوز) إلى سكر كحولي.[6] تفيد الدراسات ان الaldose reductase هو الانزيم المسؤول عن الخطوة الاولى في هذا المسار، اي ان هذا الانزيم يقوم بتحويل الجلاكتوز إلى سكر كحولي (galactitol). لكن غالاكتيتول (galactitol) لا يعد مادة مناسبة لكي يتم استهلاكها في التفاعل التالي من هذا المسار بواسطة ال بوليول ديهايدروجينيز (polyol dehydrogenase). وهكذا، يتراكم غالاكتيتول (galactitol) في أنسجة الجسم ويفرز في بول مرضى الجالاكتوسيميا، والذي بدوره يؤدي إلى العديد من الآثار السلبية.
الأكسدة إلى غالاكتونات
كما يمكن أن يخضع الجالكتوز المتراكم لتفاعل اخر وهو الأكسدة إلى غالاكتونات (galactonate). آلية تشكيل غالاكتونات لاتزال غير واضحة، ومع ذلك، تشير الدراسات الحديثة إلى أن جالاكتوز ديهايروجينيز (galactose dehydrogenase) هو المسؤول عن تحويل الجالاكتوز إلى غالاكتونولاكتون(galactonolactone)، الذي بعد ذلك تلقائيا أو إنزيميا يتحول إلى غالاكتونات. وعند تكونها، قد يدخل الغالاكتونات مسار الفوسفات بنتوز(pentose phosphate pathway). وهكذا، الأكسدة لغالاكتونات بمثابة مسار بديل لاستقلاب(metabolism) الجالاكتوز. هذا المسار التأكسدي يجعل تراكم الغالاكتونات أقل ضررا من تراكم الغالاكتيتول.
تشخيص
يتم فحص الجالاكتوسيميا عند الرضع بشكل روتيني في الولايات المتحدة، حيث يتم التشخيص في حين أن الشخص لا يزال طفلا. يظهر على الرضع الذين يعانون من ارتفاع الجالاكتوز في الدم عادة أعراض الخمول والقيء والإسهال والفشل في النمو واليرقان. أيا من هذه الأعراض ليست محددة للغالاكتوسيميا، بالتالي هناك تأخر في التشخيص. لحسن الحظ، يتم تشخيص معظم الرضع على فحص حديثي الولادة. إذا كانت أسرة الطفل حاملة لهذا المرض، يمكن للأطباء عمل اختبار قبل الولادة عن طريق أخذ عينة من السائل حول الجنين (بزل) أو من المشيمة (أخذ العينات من زغابة المشيمي أو CVS).[7]
يتم فحص الجالاكتوز في الدم (عن طريق وخز كعب الرضع)أو اختبار البول الذي يتحقق من ثلاثة إنزيمات لازمة لتغيير سكر الجالاكتوز الموجود في الحليب ومنتجات الألبان إلى الجلوكوز( السكر الذي يستخدمه جسم الإنسان للطاقة). الشخص الذي لديه غالاكتوسيميا ليس لديه واحد من هذه الانزيمات، الامر الذي يسبب مستويات عالية من الجالاكتوز في الدم أو البول.
وعادة ما يتم الكشف عن الجالاكتوز في الدم أولا من خلال فحص حديثي الولادة. يمكن أن يمتلك الأطفال المتضررين آثار خطيرة، لا رجعة فيها أو حتى يموتون في غضون أيام من الولادة من المهم أن يتم فحص المواليد الجدد للاضطرابات الأيضية دون تأخير. ويمكن الكشف عن الجالاكتوز في الدم من خلال فحص حديثي الولادة قبل تناول أي غذاء يحتوي على الجالاكتوز (يتضمن ذلك حليب الرضاعة).
لا يعتمد اكتشاف الاضطراب من خلال فحص حديثي الولادة (NBS) على تناول البروتين أو اللاكتوز، وبالتالي يجب تحديده عن طريق العينة الأولى ما لم يتم نقل دم للطفل، اي يجب أخذ عينة قبل نقل الدم.و يكون الإنزيم عرضة للتلف إذا تأخر تحليل العينة أو تعرض لدرجات حرارة عالية. إن فحص حديثي الولادة دقيق للكشف عن وجود الجالاكتوز في الدم حيث يتم استخدام اختبارين لفحص الرضع المتأثرين بالجالاكتوز في الدم اختبار بوتلر (Beutler) واختبار هيل (hill).[8] يقوم اختبار بوتلر بالفحص عن طريق تحديد مستوى انزيم الرضيع. ولذلك، فإن تناول الحليب الصناعي أو حليب الثدي لا يؤثر على الفحص، ويكون الفحص دقيق ايضا قبل تناول الجالاكتوز.
- دارتي جالاكتوسيميا: هي شكل أخف من الجالاكتوز في الدم وعادة ليس لها آثار جانبية طويلة الأمد.
أنواع
الغالاكتوز يتم تحويله إلى جلوكوز من خلال عمل ثلاثة إنزيمات يعرفوا بمسار ليلوار( Leloir pathway)، هناك أمراض متعلقة بخلل كل من هذه الانزيمات
| الأنواع | بيانات المرض | OMIM | الجين | الموقع الكروموسومي | الإنزيم | الاسم |
| النوع 1 | 5056 | 230400 | GALT | 9p13 | الجلوكتوز -1- فوسفات أورديل ترانسفيراز |
الكلاسيكوزينا الكلاسيكي |
| النوع 2 | 29829 | 230200 | GALK1 | 17q24 | غالاكتوكيناز | نقص الغالاكتوكيناز |
| النوع 3 | 29842 | 230350 | GALE | 1p36-p35 | UDP UDP إديميراس جالاكتوز epimerase | نقص جالاكتوز إمبيمراس، نقص غالاكتوز -4- إبيميراس |

الأثر
مضاعفات الجالاكتوسيميا على المدى الطويل في الدم تشمل:
- صعوبة في الكلام
- ترنح
- خلل القياس
- تناقص كثافة العظام
- فشل المبيض المبكر[9]
- الساد
العلاج
العلاج الوحيد للجالاكتوسيما هو التخلص من اللاكتوز والجالاكتوز في النظام الغذائي. حتى مع التحاليل المبكرة وإتباع النظام الغذائي، فإن بعض الأفراد الذين يعانون من الجالاكتوز في الدم (الجلاكتوزيميا) يعانون من مضاعفات على المدى الطويل، مثل: صعوبات في النطق، وصعوبات في التعلم، وتلف الأعصاب (على سبيل المثال: الهزات وغيرها)، وفشل المبيض. هذه الأعراض لا ترتبط مع دارتي جالاكتوسيميا(duarte galactosemia)، والاشخاص الذين يعانون من دارتي جالاكتوسيميا (duarte galactosemia) لا يحتاجون إلى التقيد بنظام غدائي على الاطلاق .
ومع ذلك، تؤكد البحوث صحة نظرية سبق التغاضي عنها، وهي أن دارتي جالاكتوسيميا (duarte galactosemia )قد تؤدي إلى مشاكل بتطور في اللغة عند الاطفال الذين لا يعانون من أعراض مرضية. الأطفال الرضع الذين يعانون من الجالاكتوسيميا التقليدية لا يمكن أن يرضعوا بشكل طبيعي بسبب اللاكتوز الموجود في حليب الثدي وعادة ما يتم تغذيتهم بالصويا بدل حليب الجالاكتوسيميا [10]
الجالاكتوسيميا في بعض الاحيان يتم الخلط في تشخيصها مع حساسية اللاكتوز، ولكن الجالاكتوزيميا أكثر خطورة. الاشحاص الذين يعانون من حساسية اللاكتوز يعانون من نقص إنزيم اللاكتاز بصورة مكتسبة أو موروثة ويعانون من الام البطن بعد تناول منتجات الألبان، ولكن لا يوجد آثار طويلة الأجل. في المقابل، الافراد الذين يعانون من الجالاكتوسيميا إذا استهلكوا الجالاكتوز يمكن أن يسبب ضرر دائم في أجسادهم.
مصادر
- Goppert F. (1917). "Galaktosurie nach Milchzuckergabe bei angeborenem, familiaerem chronischem Leberleiden". Klinische Wochenschrift. 54: 473–7.
- Isselbacher KJ, Anderson EP, Kurahashi K, Kalckar HM (1956). "Congenital Galactosemia, a single enzymatic block in galactose metabolism". Science. 123: 635–6. doi:10.1126/science.123.3198.635. PMID 13311516.
- "Classical Galactosaemia - Ireland's Health Service". www.hse.ie. مؤرشف من الأصل في 9 سبتمبر 2017.
- Murphy M, McHugh B, Tighe O, et al. (July 1999). "Genetic basis of transferase-deficient Galactosaemia in Ireland and the population history of the Irish Travellers". Eur. J. Hum. Genet. 7 (5): 549–54. doi:10.1038/sj.ejhg.5200327. PMID 10439960. مؤرشف من الأصل في 17 فبراير 2017.
- Galactosemia - تصفح: نسخة محفوظة April 19, 2016, على موقع واي باك مشين. The University of Utah, Genetics Science Learning Center. 2008.
- Kolatkar, Nikheel Dr. "Aldose Rudctase Inhibitors." Your Total Health. "Archived copy". مؤرشف من الأصل في 12 يوليو 200919 يناير 2011.
- Fensom AH, Benson PF, Blunt S (November 1974). "Prenatal diagnosis of galactosaemia". Br Med J. 4 (5941): 386–7. doi:10.1136/bmj.4.5941.386. PMC . PMID 4154122.
- Parris CR (August 2006). "An Overview of Expanded Newborn Screening for Inborn Errors of Metabolism" ( كتاب إلكتروني PDF ). Nutrition issues in Gastroenterology. مؤرشف من الأصل ( كتاب إلكتروني PDF ) في 20 سبتمبر 2015.
- Waggoner DD, Buist NR, Donnell GN (1990), "Long-term prognosis in galactosaemia: results of a survey of 350 cases..", J Inherit Metab Dis, 13: 802–18, PMID 1706789
- "Breastfeeding: Diseases and Conditions: Contraindicators | DNPA". www.cdc.gov. مؤرشف من الأصل في 01 مايو 2017.