رقاقة التسلسل، تسمى أيضا ChIP-seq ، هي طريقة مستخدمة لتحليل تفاعلات البروتين مع الحمض النووي . يجمع ChIP-seq بين المناعة المناعية للكروماتين (ChIP) وتسلسل الحمض النووي المتوازي بشكل كبير لتحديد مواقع الربط للبروتينات المرتبطة بالحمض النووي. يمكن استخدامه لتعيين مواقع الربط بشكل عمومي بالتحديد لأي بروتين مهم. في السابق، كان Chip-on-chip هو الأسلوب الأكثر شيوعًا المستخدم لدراسة علاقة الحمض النووي بالبروتين .
الاستخدامات
يستخدم ChIP-seq بشكل أساسي لتحديد كيفية تأثير عوامل النسخ والبروتينات الأخرى المرتبطة بالكروماتين على آليات التي تؤثر على النمط الظاهري . يعد تحديد كيفية تفاعل البروتينات مع الحمض النووي لتنظيم التعبير الجيني أمرًا ضروريًا لفهم العديد من العمليات البيولوجية وحالات الأمراض. هذه المعلومات الجينية مكملة للنمط الجيني وتحليل التعبير. ينظر إلى تقنية ChIP-seq حاليًا كبديل لشريحة Chip التي تتطلب مصفوفة بروتين دقيقة. يقدم هذا بالضرورة بعض التحيز، حيث يقتصر المصفوفة على عدد محدد من التحقيقات. بالمقابل، يُعتقد أن التسلسل أقل تحيزًا، على الرغم من أن التحيز التسلسلي لتقنيات التسلسل المختلفة لم يتم فهمه بشكل كامل حتى الآن.
مواقع الحمض النووي المحددة في التفاعل الجسدية المباشر مع عوامل النسخ والبروتينات الأخرى يمكن عزلها عن طريق الترسيب المناعي الكروماتين . تنتج ChIP مكتبة مواقع الحمض النووي المستهدفة المرتبطة ببروتين مهم في الجسم الحي . تُستخدم تحليلات التسلسل المتوازي بشكل كبير بالاقتران مع قواعد بيانات تسلسل الجينوم الكامل لتحليل نمط تفاعل أي بروتين مع الحمض النووي، [1] أو نمط أي تعديلات كروماتينية جينية. يمكن تطبيق ذلك على مجموعة البروتينات والتعديلات القادرة للحصول على chip ، مثل عوامل النسخ والبوليمرات والآلات النسخية والبروتينات الهيكلية وتعديلات البروتين وتعديلات الحمض النووي . [2] وكبديل للاعتماد على أجسام مضادة محددة، وتم تطوير طرق مختلفة للعثور على مجموعة شاملة لجميع المناطق التنظيمية النشطة في جسيم نووي المستنفدة للنيوكليوم أو النيوكليوسوم في الجينوم مثل DNase و FAIRE seq .
سير العمل رقاقة التسلسل
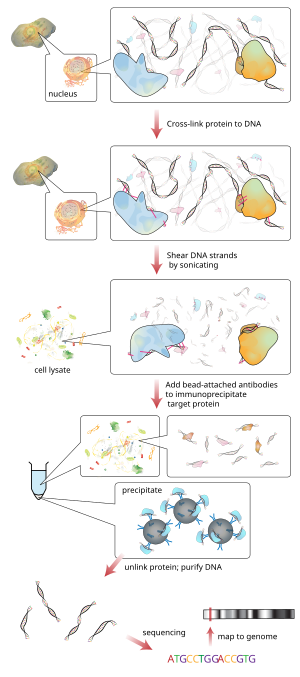
رقاقة
ChIP هي وسيلة قوية لإثراء انتقائي لتسلسل الحمض النووي المرتبطة ببروتين معين في الخلايا الحية. ومع ذلك، فإن الاستخدام الواسع النطاق لهذه الطريقة كان محدودا بسبب عدم وجود طريقة قوية بما فيه الكفاية لتحديد جميع تسلسل الحمض النووي المحسن تعمل عملية ChIP على إثراء مجموعات معينة من بروتين الحمض النووي المتشابك باستخدام جسم مضاد ضد البروتين المستهدف للحصول على وصف جيد لبروتوكول مختبر الرطب من chip، انظر ChIP-on-chip . ثم تضاف محولات أوليغنيكليوتيد إلى الامتدادات الصغيرة من الحمض النووي التي كانت مرتبطة بالبروتين المستهدف لتمكين التسلسل المتوازي على نطاق واسع.
التسلسل
بعد اختيار الحجم، يتم تسلسل جميع شظايا ChIP-DNA الناتجة في وقت واحد باستخدام منظم الجينوم. يمكن لمسار تسلسلي واحد أن يبحث عن الارتباطات على نطاق الجينوم بدقة عالية، مما يعني أنه يمكن تحديد الميزات بدقة على الكروموسومات. على النقيض من ذلك، تتطلب رقاقة Chips مجموعات كبيرة من صفائف التجانب لخفض الدقة.
هناك العديد من طرق التسلسل الجديدة المستخدمة في هذه الخطوة التسلسل. يمكن لبعض التقنيات التي تقوم بتحليل التسلسلات استخدام التضخيم العنقودي لشظايا ChIP DNA ذات محول في الركيزة الصلبة لخلية التدفق لإنشاء مجموعات من حوالي 1000 نسخة نسيلية لكل منها. يتم ترتيب مجموعة الكثافة العالية الناتجة من مجموعات القوالب على سطح خلية التدفق بواسطة برنامج تحليل الجينوم. يخضع كل قالب مجموعة التسلسل تلو التوازي في موازاة استخدام نيوكليوتيدات فاصل عكسي المسمى fluorescently. يتم تسلسل القوالب كل قاعدة على حدة أثناء كل قراءة. بعد ذلك، يقوم برنامج جمع وتحليل البيانات بمحاذاة تسلسل العينات مع تسلسل جينومي معروف لتحديد شظايا ChIP-DNA المطلوبة .
حساسية
تعتمد حساسية هذه التقنية على عمق المدى التسلسلي (أي عدد علامات التسلسل المعين) وحجم الجينوم وتوزيع العامل المستهدف. يرتبط عمق التسلسل مباشرة بالتكلفة. إذا كان من الضروري تعيين روابط وفيرة في جينومات كبيرة بحساسية عالية، فستكون التكاليف مرتفعة حيث ستكون هناك حاجة إلى عدد كبير للغاية من علامات التسلسل. وهذا يتناقض مع رقاقة Chip حيث لا ترتبط التكاليف بالحساسية.
وعلى عكس طرق ChIP المستندة إلى مصفوفة البروتين الدقيقة ، فان دقة اختبار ChIP-seq لا تقتصر على تباعد التحقيقات المحددة مسبقًا. من خلال دمج عدد كبير من القراءات القصيرة، يتم الحصول على توطين موقع الربط الدقيق للغاية. مقارنةً برقاقة ChIP ، يمكن استخدام بيانات ChIP-seq لتحديد موقع الربط ضمن عشرات قليلة من الأزواج الأساسية لموقع ربط البروتين الفعلي. تعد كثافات العلامات في مواقع الربط هي مؤشرًا جيدًا على تقارب ارتباط البروتين بالحمض النووي، [3] مما يجعل من السهل تحديد ومقارنة الارتباطات المرتبطة للبروتين بمواقع الحمض النووي المختلفة. [4]
البحوث الحالية
رابطة الحمض النووي (الحالة 1): تم استخدام ChIP-seq لدراسة أهداف STAT1 في خلايا HLA S3. ثم تمت مقارنة أداء ChIP-seq بطرق تفاعل البروتين والحمض النووي البديلة في ChIP-PCR و ChIP-chip. [5]
بنية نووية للمروجين: باستخدام ChIP-seq ، تم تحديد أن جينات الخميرة يبدو أنها تحتوي على أقل منطقة محفِّزة خالية من النيوكليوسوم تبلغ 150 نقطة أساس والتي يمكن أن يبدأ فيها بوليميريز الحمض النووي الريبي النسخ. [6]
الحفاظ على عامل النسخ: تم استخدام ChIP-seq لمقارنة حفظ الـ TFs في الدماغ الأمامي وأنسجة القلب في الفئران الجنينية. حدد المؤلفون والتحقق من صحة وظائف القلب من معززات النسخ، وقرروا أن معززات النسخ للقلب هي أقل تحفظا من تلك الموجودة في الدماغ الأمامي خلال نفس المرحلة التنموية. [7]
ChIP-seq على نطاق الجينوم: اكتمل تسلسل ChIP على الدودة (C. elegans )لاستكشاف مواقع الربط على نطاق الجينوم التي تضم 22 عاملاً من النسخ. تم تعيين ما يصل إلى 20 ٪ من الجينات المرشحة المشروحة لعوامل النسخ. تم تعيين العديد من عوامل النسخ لمناطق الحمض النووي الريبي غير المشفرة وقد تخضع لمتغيرات تطورية أو بيئية. كما تم تحديد وظائف بعض عوامل النسخ. بعض عوامل النسخ تنظم الجينات التي تتحكم في عوامل النسخ الأخرى. لا يتم تنظيم هذه الجينات من قبل عوامل أخرى. تعمل معظم عوامل النسخ كأهداف ومنظمين لعوامل أخرى، مما يدل على وجود شبكة من التنظيم . [8]
الاستدلال على الشبكة التنظيمية: تم عرض إشارة Chip-seq لتعديل هيستون على أنها أكثر ارتباطًا بزخارف عامل النسخ عند المروجين مقارنة بمستوى الحمض النووي الريبي. [9] ومن هنا اقترح المؤلف أن استخدام تعديل هيستون يوفر ChIP-seq استدلالًا أكثر موثوقية للشبكات التنظيمية الجينية مقارنةً بالطرق الأخرى القائمة على التعبير.
يقدم ChIP-seq بديلاً Chip-chip : تتمتع بيانات STAT1 التجريبية بدرجة عالية من التشابه مع النتائج التي تم الحصول عليها بواسطة Chip-chip لنفس النوع من التجربة، مع أكثر من 64٪ من القمم في المناطق الجينية المشتركة. نظرًا لأن البيانات عبارة عن قراءة تسلسلية، تقدم ChIP-seq خطًا للتحليل السريع (طالما يتوفر تسلسل جينوم عالي الجودة لرسم الخرائط للقراءة، ولا يحتوي الجينوم على محتوى متكرر يربك عملية التعيين) القدرة على اكتشاف الطفرات في تسلسل مواقع الربط، والتي قد تدعم مباشرة أي تغييرات ملحوظة في ربط البروتين وتنظيم الجينات.
التحليل الحسابي
كما هو الحال مع العديد من أساليب التسلسل عالية الإنتاجية، تقوم ChIP-seq بإنشاء مجموعات بيانات كبيرة للغاية، والتي تتطلب طرق التحليل الحسابية المناسبة. للتنبؤ بمواقع ارتباط الحمض النووي من بيانات ChIP-seq ، تم تطوير طرق الاتصال القصوى . الطريقة الأكثر شعبية هو MACS الذي يصمم تجريبياً حجم التحول لعلامات ChIP-Seq ويستخدمه لتحسين الدقة المكانية لمواقع الربط المتوقعة. [10]
هناك مشكلة حسابية أخرى ذات صلة وهي النداء الذروي التفاضلي، الذي يحدد اختلافات كبيرة في إشارات تشيب-ساق من ظروف بيولوجية متميزة. الجزء المتصل بالذروة التفاضلية يشتمل على اثنين من إشارات ChIP-seq ويحدد الذروة التفاضلية باستخدام نماذج Hidden Markov . أمثلة للمتصلين الذروة التفاضلية على مرحلتين هي ChIPDiff [11] و ODIN. [12]
مقالات ذات صلة
- Chip on chip
- رقاقة -PET
- رقاقة-PCR
طرق مماثلة
- تسلسل CUT & RUN ، انقسام مستهدف للأجسام المضادة بواسطة nuclease micrococcal بدلاً من ChIP ، مما يسمح بزيادة نسبة الإشارة إلى الضوضاء أثناء التسلسل.
- Sono-Seq ، مطابقة لـ ChIP-Seq لكن تخطي خطوة مناعي.
- HITS-CLIP [13] [14] (وتسمى أيضًا CLIP-Seq ) ، لإيجاد تفاعلات مع RNA بدلاً من DNA.
- PAR-CLIP ، طريقة أخرى لتحديد مواقع الربط لبروتينات ربط الحمض النووي الريبي الخلوية (RBPs).
- RIP-Chip ، نفس الهدف والخطوات الأولى، ولكن لا يستخدم طرق الربط المتقاطع ويستخدم ميكروأري بدلاً من التسلسل
- SELEX ، طريقة لإيجاد تسلسل ملزم بالإجماع
- المنافسة رقاقة، لقياس ديناميات استبدال النسبية على الحمض النووي.
- ChiRP-Seq لقياس الحمض النووي الريبي المرتبط بالحمض النووي الريبي والبروتينات.
- يستخدم ChIP-exo علاج نوكلياز لتحقيق ما يصل إلى قرار واحد الزوج
- ChIP-nexus نسخة محسنة من ChIP-exo لتحقيق ما يصل إلى دقة زوج واحد.
- يستخدم DRIP-seq جسمًا مضادًا S9.6 لترسيب الهجينة DND: RNA الثلاثية تقطعت بهم السبل وتسمى R-loops.
- TCP-seq ، طريقة مشابهة بشكل أساسي لقياس ديناميات ترجمة mRNA.
- Calling Cards ، يستخدم transposase لتحديد التسلسل حيث يرتبط عامل النسخ. [15]
المراجع
- Johnson, DS; Mortazavi, A; et al. (2007). "Genome-wide mapping of in vivo protein–DNA interactions" ( كتاب إلكتروني PDF ). Science. 316 (5830): 1497–1502. doi:10.1126/science.1141319. PMID 17540862. مؤرشف من الأصل ( كتاب إلكتروني PDF ) في 24 يوليو 2018.
- ( كتاب إلكتروني PDF ) https://web.archive.org/web/20200103004636/https://www.illumina.com/Documents/products/datasheets/datasheet_chip_sequence.pdf. مؤرشف من الأصل ( كتاب إلكتروني PDF ) في 3 يناير 2020.
- Jothi et al. (2008) Genome-wide identification of in vivo protein–DNA binding sites from ChIP-seq data. Nucleic Acids Res 36(16) 5221–5231.
- Bernstein, BE; et al. (2005). "Genomic maps and comparative analysis of histone modifications in human and mouse". Cell. 120 (2): 169–181. doi:10.1016/j.cell.2005.01.001. PMID 15680324.
- Robertson, G; et al. (2007). "Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing". Nature Methods. 4 (8): 651–657. doi:10.1038/nmeth1068.
- Schmid; et al. (2007). "ChIP-Seq Data reveal nucleosome architecture of human promoters". Cell. 131 (5): 831–832. doi:10.1016/j.cell.2007.11.017.
- Blow, M. J.; McCulley, D. J.; Li, Z.; Zhang, T.; Akiyama, J. A.; Holt, A.; Plajzer-Frick, I.; Shoukry, M.; Wright, C. (2010). "ChIP-seq identification of weakly conserved heart enhancers". Nature Genetics. 42 (9): 806–810. doi:10.1038/ng.650. PMID 20729851.
- Niu, W.; Lu, Z. J.; Zhong, M.; Sarov, M.; Murray, J. I.; Brdlik, C. M.; Janette, J.; Chen, C.; Alves, P. (2011). "Diverse transcription factor binding features revealed by genome-wide ChIP-seq in C. elegans". Genome Research. 21 (2): 245–254. doi:10.1101/gr.114587.110.
- Vibhor Kumar, Masafumi Muratani, Nirmala Arul Rayan, Petra Kraus, Thomas Lufkin, Huck Hui Ng and Shyam Prabhakar, Uniform, optimal signal processing of mapped deep-sequencing data, Nature biotechnology, 2013
- Zhang, Y; Liu, T; Meyer, CA; Eeckhoute, J; Johnson, DS; Bernstein, BE; Nusbaum, C; Myers, RM; Brown, M (2008). "Model-based analysis of ChIP-Seq (MACS)". Genome Biol. 9 (9): R137. doi:10.1186/gb-2008-9-9-r137. PMID 18798982.
- Xu, Sung; Wei; Lin (28 July 2008). "An HMM approach to genome-wide identification of differential histone modification sites from ChIP-seq data". Bioinformatics. 24 (20): 2344–2349. doi:10.1093/bioinformatics/btn402. PMID 18667444.
- Allhoff, Costa; Sere; Chauvistre; Lin; Zenke (24 October 2014). "Detecting differential peaks in ChIP-seq signals with ODIN". Bioinformatics. 30 (24): 3467–3475. doi:10.1093/bioinformatics/btu722.
- "HITS-CLIP yields genome-wide insights into brain alternative RNA processing". Nature. 456 (7221): 464–9. November 2008. doi:10.1038/nature07488. PMID 18978773.
- Darnell RB (2010) HITS-CLIP: panoramic views of protein-RNA regulation in living cells. Wiley Interdiscip Rev RNA. 1):266-86. doi:10.1002/wrna.31
- Wang, H.; Mayhew, D.; Chen, X.; Johnston, M.; Mitra, R. D. (6 April 2011). "Calling Cards enable multiplexed identification of the genomic targets of DNA-binding proteins". Genome Research. 21 (5): 748–755. doi:10.1101/gr.114850.110.
روابط خارجية
- كتالوج ReMap : تحليل متكامل وموحد لـ ChIP-Seq للعناصر التنظيمية من مجموعات بيانات ChIP-seq +2800 ، مما يوفر فهرسًا يتكون من 80 مليونًا من 485 منظم نسخ. يوصف التحليل بالتفصيل في هذه الورقة . [1]
- قاعدة بيانات ChIPBase : قاعدة بيانات لاستكشاف خرائط ربط عامل النسخ من بيانات ChIP-Seq . يوفر مجموعة بيانات ChIP-Seq الأكثر شمولًا لأنواع وظروف الخلايا / الأنسجة المختلفة.
- GeneProf أداة تحليل وقاعدة بيانات : GeneProf هي بيئة تحليل سهلة الاستخدام ويمكن الوصول إليها بحرية لبيانات ChIP-seq و RNA-seq ، وتأتي مع قاعدة بيانات كبيرة من التجارب العامة الجاهزة للتحليل، على سبيل المثال لتعديلات ملزمة عوامل النسخ وتاريخ هيستون. يتم وصف قاعدة البيانات بالتفصيل في هذه الورقة .
- الفرق الذروة الدعوة : دروس لذروة التفاضلية الدعوة مع ODIN.
- التحليل المعلوماتي الحيوي لبيانات ChIP-seq : تم توضيح الإرشادات العملية للتحليل الشامل لبيانات ChIP-seq في ** * هذه الورقة . [2]
- KLTepigenome : كشف التباين المترابط في مجموعات البيانات اللاجينية باستخدام تحويل Karhunen-Loeve.
- SignalSpider : أداة لاكتشاف النمط الاحتمالي في ملفات تعريف إشارة ChIP-Seq متعددة
- FullSignalRanker : أداة للانحدار وتنبؤ الذروة على ملفات تعريف إشارة ChIP-Seq متعددة
Translated by:Eng.Esraa Alquraan and Dr. Ahmad omari
موسوعات ذات صلة :
- Chèneby, Jeanne; Gheorghe, Marius; Artufel, Marie; Mathelier, Anthony; Ballester, Benoit (2018). "ReMap 2018: an updated atlas of regulatory regions from an integrative analysis of DNA-binding ChIP-seq experiments". Nucleic Acids Research. 46: D267–D275. doi:10.1093/nar/gkx1092.
- Bailey, T; Krajewski, P; Ladunga, I; Lefebvre, C; Li, Q; et al. (2013). "Practical Guidelines for the Comprehensive Analysis of ChIP-seq Data". PLoS Comput Biol. 9 (11): e1003326. doi:10.1371/journal.pcbi.1003326.