
Le chromosome X est l'un des deux chromosomes sexuels de l'être humain et de certains animaux (l'autre chromosome sexuel étant le chromosome Y). Il fait partie du système XY de détermination sexuelle.
Caractéristiques du chromosome X humain
- Nombre de paires de base : 154 824 264
- Nombre de gènes : 931
- Nombre de gènes connus : 766
- Nombre de pseudogènes : 380
- Nombre de variations des nucléotides (polymorphismes nucléotidiques) : 320 997
- L'image ci-dessous illustre la richesse génétique du chromosome X à comparer avec le chromosome Y. La zone en vert localise le centre de l'inactivation de l'X.
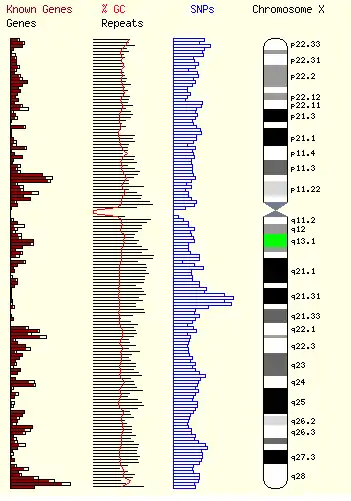
Inactivation du chromosome X ou lyonisation
Les hommes ont un seul chromosome X et les femmes deux chromosomes X. Ce chromosome X supplémentaire n'aboutit pas à une « surproduction » des protéines codées par ce chromosome. Par exemple, les facteurs de coagulations comme le facteur VIII est codé par un gène situé sur le chromosome X. Les femmes ne possèdent pas pour autant deux fois plus de facteur VIII.
Il existe donc une inactivation de l'un des deux chromosomes qui prend une forme condensée et visible, le corpuscule de Barr ou corps de Barr. Ce « corpuscule » se situe généralement dans le noyau de la cellule, sous la membrane interne de l'enveloppe nucléaire.
Le processus d’inactivation est aléatoire, se faisant au stade précoce du blastocyste, et se transmet d'une génération cellulaire à l'autre. La moitié des cellules de la femme sont sous la dépendance du chromosome X paternel et l'autre moitié sous la dépendance du chromosome X maternel. Les tissus somatiques féminins sont des mosaïques génétiques.
Ce processus d’inactivation a été soupçonné par Mary F. Lyon en 1961 et est aussi parfois appelé « lyonisation ».
Pour comprendre certains mécanismes, il est important de savoir que le chromosome X est inactivé par un centre d'inactivation (site du chromosome X) :
Quand celui-ci sera inactivé, des enzymes vont aller sur le centre d'inactivation et se propager le long du chromosome pour inactiver la plupart des gènes.
Par conséquent, si un des deux chromosomes X subit une translocation avec, par exemple, le chromosome 21 alors, dans les cellules où le chromosome X inactivé est celui touché par la translocation, une partie du chromosome 21 sera inactivée tandis qu'une autre partie du chromosome X ne le sera pas.
Anomalies chromosomiques décrites au niveau du chromosome X
- Syndrome de Klinefelter : 47,XXY
- Syndrome triple X : 47,XXX ou trisomie X
- Syndrome de Turner : 45,X
- Duplication du gène MECP2
- Délétions impliquant le gène SHOX
- Ichtyose lié à l'X (X-linked_ichthyosis (en)) par délétion du gène STS
- Incontinentia pigmenti due à la mutation du gène nemo
Gènes localisés sur le chromosome X
De nombreux gènes situés sur le chromosome X sont en cause dans des retards mentaux, ce qui pourrait expliquer, entre autres, qu'il y ait davantage de garçons porteurs d'un handicap mental que de filles[alpha 1]. Par contre, l'incontinentia pigmenti est une pathologie essentiellement féminine.
| gène | nom | locus | Nom des syndromes associés | |
| FMR1 | Xq27.3 | Syndrome de l'X fragile (site FRAXA) | ||
| ARX | Xp22.1 | Syndrome de Partington, syndrome XLAG (X-linked lissencephaly with abnormal genitalia) ou syndrome de Berry-Kravis, Syndrome de Proud, Spasmes infantiles liés à l’X (syndrome de West) | ||
| MECP2 | Xq28 | Syndrome de Rett, Syndrome de la duplication du gène MECP2, PPM-X (psychosis,pyramidal signs,
macroorchidism), Retard mental avec spasticité (Meloni) | ||
| nemo | Xq28 | code la protéine NEMO (IKKg). Sa mutation est responsable de l'Incontinentia pigmenti | ||
| CDKL5 | STK9 | Xp22 | Syndrome de Rett variant, Crises partielles précoces +/- Spasmes infantiles (syndrome de West)
ISSX | |
| PQBP1 | Xp11.23 | Renpenning syndrome, Sutherland syndrome, Golabi-Ito-Hall syndrome | ||
| XNP | Xq13.3 | Syndrome ATR-X (alpha thalassémie RM lié à l’X) Syndrome de Juberg-Marsidi, Syndrome de Chudley-Lowry,Syndrome de Smith-Fineman-Myers, Syndrome de Carpenter-Waziri, Syndrome de Holmes-Gang, Syndrome de Martinez | ||
| JARID1C/ SMCX | Xp11.22 | microcéphalie | ||
| DCX | Xq23 | Syndrome SCLH (subcortical laminar heterotopia), XLIS | ||
| OPHN1 | Xq12 | Retard mental lié à l’X avec hypoplasie cérébelleuse | ||
| PHF8 | Xp11.22 | Syndrome de Siderius-Hamel | ||
| PHF6 | Syndrome de Borjeson-Forssman-Lehmann | |||
| FMR2 | X fragile site FRAXE (RM non spécifique léger) | |||
| GDI1 | ||||
| AGTR2 | ||||
| ZNF41, ZNF 81, ZNF 674 | Gènes des zinc finger protéines | |||
| ILIRAPL1 | Interleukine 1 receptor accesory proteine like 1 ; | |||
| TM4SF2/TSPAN7 tétraspanine 7 | ||||
| DLG3 | ||||
| FTSJ1 | ||||
| FACL4 | ||||
| PAK3 | ||||
| MECP2 | ||||
| SLC6A8 | Xq28 | Déficit en transporteur de la créatine | ||
| PLS3 | Plastine 3 | |||
| FUNDC1 | ||||
| Tous les autres… | ||||
Source: Pr des Portes - Déc 2006
Maladies localisées sur le chromosome X
- La nomenclature utilisée pour localiser un gène est décrite dans l'article de celui-ci
- Les maladies en rapport avec des anomalies génétiques localisées sur le chromosome X sont :
| Maladies localisées sur le chromosome X | |||||
|---|---|---|---|---|---|
| Pathologie | Transmission | O.M.I.M | Locus | Gène | Protéine codée par le gène |
| Bras long | |||||
| Dysplasie frontométaphysaire | Q28 | ||||
| Syndrome oto-palato-digital type I et type II | Q28 | ||||
| Syndrome de Melnick-Needles | Q28 | ||||
| Hétérotopie nodulaire héréditaire liée à l' X | Q28 | ||||
| Dystrophie musculaire d'Emery-Dreifuss | Q28 | ||||
| Adrénoleucodystrophie liée à l'X | Q28 | ||||
| Syndrome CHILD | Q28 | ||||
| Hydrocéphalie liée à l'X | Q28 | ||||
| Syndrome M.A.S.A | Q28 | ||||
| Paraplégie spastique familiale de type 1 | Q28 | ||||
| Syndrome de Rett | Q28 | ||||
| Hémophilie A | Récessive liée à l'X | 306700 | Q28 | ||
| Daltonisme | Q28 | ||||
| Myopathie congénitale myotubulaire | Récessive liée à l'X | 310400 | q28 | MTM1 | |
| déficience intellectuelle associée au site fragile FRAXE | Dominante liée à l'X | 309548 | Q28 | AFF2 (en) | |
| Syndrome de l'X fragile | Q27.3 | ||||
| Hémophilie B | Récessive liéeà l'X | 306900 | q27.1-q27.2 | ||
| Syndrome de Borjeson-Forssman-Lehmann | Q26.3 | ||||
| Syndrome de Lowe | Q26.1 | ||||
| Syndrome de Lesch-Nyhan | Q26 | ||||
| Q26 | |||||
| Maladie lymphoproliférative liée à l'X | Q25 | ||||
| Q25 | |||||
| Q23.1 | |||||
| Q23 | |||||
| Albinisme oculaire lié à l'X | Q22.3 | ||||
| Syndrome d'Alport | Q22.3 | ||||
| Maladie de Fabry | Q22 | ||||
| Maladie de Pelizaeus-Merzbacher | Q22 | ||||
| Paraplégie spastique familiale type 2 | Q22 | ||||
| Syndrome de Mohr-Tranebjaerg | Q22 | ||||
| Agammaglobulinémie liée au sexe | Récessive liée à l'X | 300300 | q21.3-q22 | BTK | |
| Myopathie de Duchenne | Q21.2 | ||||
| Q21 | |||||
| Syndrome de la corne occipitale | Q13.3 | ||||
| Maladie de Menkès | Q13.3 | ||||
| Anémie sidéroblastique liée à l'X avec ataxie | Récessive liée à l'X | 301310 | q13.1-q13.3 | ABCB7 | |
| Dystonie avec parkinsonisme liée à l'X | Q13.1 | ||||
| Maladie de Charcot-Marie-Tooth lié à l'X | Q13 | ||||
| Thalassémie alpha liée à l'X avec retard mental | Q13 | ||||
| Q13 | |||||
| Q12 | |||||
| Syndrome d'insensibilité aux androgènes | Q11 | ||||
| Maladie de Kennedy | Q11 | ||||
| Bras court | |||||
| p11 | |||||
| Chondrodystrophie calcifiante congénitale | |||||
| Syndrome d'Aarskog | p11.21 | ||||
| Syndrome de Cornelia de Lange | 300590 | p11.21 | SMC1L1 | ||
| Syndrome Homme XX | P11.3 | ||||
| Syndrome oculo-facio-cardio-dentaire | P11.4 | ||||
| Syndrome de McLeod | Récessive | 314850 | P21.1 | XK | |
| Déficit en Glycérol Kinase | p21.3-p21.2 | Glycérol kinase | |||
| Hypoplasie Congénitale des Surrénales | p21.3-p21.2 | Gène DAX-1 | |||
| Syndrome d'Aicardi | P22 | ||||
| Syndrome d’Opitz lié à l’X | |||||
| Syndrome de Coffin-Lowry | P22.1 | ||||
Liens externes
Earlier versions of this article contain material from the National Library of Medicine (https://www.nlm.nih.gov/copyright.html) , a part of the National Institutes of Health (USA,) which, as a US government publication, is in the public domain.
- Ressources relatives à la santé :
- FMA
- (en) Medical Subject Headings
- (cs + sk) WikiSkripta
- Notices dans des dictionnaires ou encyclopédies généralistes :
- (en) Ensemble Genome Browser
- (en) Online Mendelian Inheritance in Man (TM), OMIM (TM), université Johns-Hopkins, Baltimore, Maryland.
- (en) L. Carrel L, HF. Willard, « X-inactivation profile reveals extensive variability in X-linked gene expression in females », dans Nature 434 (2005), 400-404. [(en) lire en ligne]
Notes et références
Notes
- ↑ un allèle récessif porté par le chromosome X s'exprime chez l'homme qui n'a qu'un de ces chromosomes, alors qu'il a peu de chance de s'exprimer chez la femme où il faudrait avoir la mutation sur les deux chromosomes X.