| Causes | Génétique |
|---|---|
| Symptômes | Œdème, ictère, modification de la personnalité (en), ongles bleus (en) et signe du panda géant (en) |
| Médicament | Triéthylènetétramine, pénicillamine et triéthylènetétramine |
|---|---|
| Spécialité | Endocrinologie |
| CISP-2 | A91 |
|---|---|
| CIM-10 | E83.0 |
| CIM-9 | 275.1 |
| OMIM | 277900 |
| DiseasesDB | 14152 |
| MedlinePlus | 000785 |
| eMedicine |
183456 neuro/570ped/2441 |
| MeSH | D006527 |
| GeneReviews | |
| Patient UK | Wilsons-disease-pro |
![]() Mise en garde médicale
Mise en garde médicale
La maladie de Wilson est une maladie génétique secondaire liée à une accumulation de cuivre dans l'organisme et se manifestant par des atteintes du foie et du système nerveux.
Historique
La maladie a été décrite pour la première fois par Kinnier Wilson en 1912[1]. Le gène responsable a été identifié à la fin des années 1980.
Avant les années 1950, elle était constamment mortelle. Les premiers traitements sont apparus vers cette époque, d'abord le dimercaprol, puis la pénicillamine en 1956[2], la trientine à la fin des années 1960. L'utilisation du zinc date des débuts des années 1960.
Épidémiologie
La fréquence de cette maladie est très faible : 1 sur 30 000 à 100 000 naissances[3].
Mécanisme
Elle est due à une anomalie d'un gène qui intervient dans le métabolisme du cuivre. La maladie de Wilson est une maladie génétique à transmission autosomique récessive : les deux parents étant simplement porteurs sains de ce gène.
Le gène anormal est situé sur le chromosome 13. 1 % de la population générale est porteur du gène anormal (porteurs sains hétérozygotes). En France, 1 000 à 1 500 personnes sont atteintes de cette maladie. Il est principalement exprimé dans le foie et les reins.
Ce gène, nommé ATP7B, code une protéine trans-membranaire de type ATPase, appelée ATP7B, intervenant dans le transport intra et extra cellulaire du cuivre, permettant de réguler la concentration de ce métal et son excrétion dans la bile. Si la protéine est déficiente, le métal s'accumule alors à l'intérieur des cellules.
Il existe près de 300 mutations décrites de ce gène mais seule une poignée de ces dernières est responsable de l'essentiel des cas de maladie de Wilson. Le type de mutation dépend également de l'origine ethnique du patient[3]. En Europe, le type H1069Q est prédominant.
Sémiologie
Bien que les déficits biochimiques soient présents dès la naissance, les symptômes cliniques apparaissent rarement avant l'âge de cinq ans. Les manifestations de cette maladie sont en rapport avec l’accumulation du cuivre dans le foie (jusqu’à 20 fois les taux normaux), le cerveau et l’œil (le cuivre est transporté par le sang vers ces organes). La céruloplasmine plasmatique est typiquement en dessous de 30 % de sa valeur normale.
L'âge auquel apparaissent les premiers symptômes dépend des organes atteints. Environ la moitié des patients (40-50 %) manifestent d'abord des symptômes hépatiques (foie) et la moitié (40-50 %) des symptômes neurologiques. L'âge moyen des symptômes hépatiques est de 10 à 14 ans, celui des symptômes neurologiques de 19 à 22 ans. Toutefois des enfants adolescents de moins de 14 ans ont subi des troubles neurologiques et hépatiques sévères.
Foie
Rarement, la maladie se présente sous forme d'hépatite fulminante gravissime avec troubles de la conscience, insuffisance rénale aigüe et saignements diffus par trouble de la coagulation. Le plus souvent la maladie se présente sous forme d'une Hépatite chronique active, voire au stade d'une cirrhose.
L'atteinte du foie précède en règle générale l'atteinte neurologique de quelques années. Mais les troubles neurologiques (notamment chez l’enfant) peuvent se présenter avant l’atteinte du foie (aphasie, troubles du langage, troubles de motricité)[4].
Cerveau
Les signes neurologiques ou psychiatriques concernent près de 50 % des patients atteint de la maladie de Wilson[3]. L'imagerie par résonance magnétique (IRM) montre des lésions de plusieurs structures cérébrales, même en l'absence de tout signe clinique et l'importance de celles-ci semble corrélée avec le degré d'avancement de la maladie[5].
Psychiatrie
Des troubles psychiques peuvent apparaître les premiers et consistent en un certain désintérêt de l’activité scolaire ou professionnelle, des modifications du caractère avec une hyperémotivité avec une grande labilité de l’humeur, des syndromes dépressifs, des états de psychose. Ils contrastent avec la conservation des fonctions intellectuelles.
Neurologie
L'atteinte peut comporter :
- une ataxie ;
- des dyskinésies ;
- une rigidité semblable à celle de la maladie de Parkinson ;
- des tremblements.
Œil
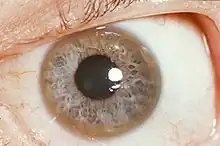
Les anneaux de Kayser-Fleischer sont typiques de la maladie, consistant en une discrète coloration marron clair du pourtour extérieur de la cornée secondaire aux dépôts de cuivre dans la membrane de Descemet. Un aspect semblable (mais sans déposition métallique) peut être vu cependant dans d'autres formes de cirrhose.
Une cataracte est une complication classique. Elle se caractérise par un aspect étoilé.
Autres atteintes
Il peut exister :
- des atteintes osseuses ou articulaires ou ostéoarticulaires[6] et un lien avec la spondylarthrite ankylosante a été évoqué[7] ;
- une atteinte rénale (rare), comprenant une acidose tubulaire ou une lithiase rénale ;
- une atteinte cardiaque (rare, du moins dans son expression clinique). Des cardiomyopathie ainsi que des troubles du rythme cardiaque ont été décrits.
Un signe classique est l'aspect bleuté de la lunule des ongles.
Diagnostic
Le taux de céruléoplasmine sanguine (protéine comportant du cuivre en son sein) est bas, voire effondré.
Le dosage du contenu en cuivre du tissu hépatique est un bon examen (il est augmenté lors d'une maladie de Wilson) mais nécessite de recourir à une biopsie du foie. Dans quelques cas, cependant, cette concentration peut être normale (notamment en cas de fibrose hépatique évoluée).
L'anneau de Kayser-Fleischer est un indice qui peut être déterminant dans le diagnostic de la maladie, mais ces anneaux ne sont pas forcément présents en cas de Wilson. La présence d'un dépôt bleuté à la base des ongles peut également être un bon indicateur. L'excrétion urinaire en cuivre est augmentée lors de la maladie, mais il existe un certain nombre de faux positifs. Sa mesure peut être sensibilisée chez l'enfant par l'administration de pénicillamine[8].
Ces examens peuvent être perturbés chez les hétérozygotes (ne présentant donc pas la maladie puisqu'elle est récessive).
La prise de sang permet de faire également le diagnostic des formes pré-cliniques c’est-à-dire avant l’apparition des signes de la maladie qui peut mettre des dizaines d’années avant de se manifester.
En règle générale, l'identification génétique n'est pas indispensable dans les cas typiques pour en faire le diagnostic. Il peut être utile dans les cas douteux.
Traitement
L'objectif du traitement est d'éradiquer la toxicité des dépôts de cuivre. Le traitement doit être suivi à vie. Il ne doit jamais être interrompu. Il consiste à prendre des médicaments chélateurs qui diminuent l’absorption de cuivre dans l’organisme, ou à augmenter l'excrétion du métal. Le traitement doit être soumis à une surveillance périodique de façon à repérer l'apparition d'effets secondaires indésirables.
Différents types de médicaments sont employés :
- la D-pénicillamine augmente l'excrétion urinaire du cuivre. Elle a une efficacité reconnue mais ses effets secondaires tendent à la faire remplacer par d'autres molécules ;
- la trientine qui est un chélateur du cuivre, souvent mieux toléré que la pénicillamine, mais moins efficace sur l'évolution neurologique[9] ;
- le tétrathiomolybdate d'ammonium , absorbé avec l'alimentation, se fixe avec les ions cuivre dans le tube digestif, empêchant leur absorption ;
- le zinc active une enzyme, la métallothionéine, qui va fixer le cuivre dans les cellules de la paroi de l'intestin (entérocytes) empêchant le passage de cet ion dans la circulation sanguine.
Un régime pauvre en cuivre est recommandé : il est conseillé d'éviter les champignons, les fruits de mer, le foie, les arachides ainsi que le chocolat.
Dans les cas graves d'hépatites fulminantes ou dans les atteintes graves essentiellement hépatique, une transplantation hépatique peut être proposée.
Conseil génétique
Il faut procéder à un dépistage de la maladie chez tous les frères et sœurs d'un patient chez qui on vient de diagnostiquer la maladie de Wilson. La maladie étant de transmission récessive, la probabilité qu'un frère ou qu'une sœur soit atteinte est de 1/4. Toute la difficulté consiste à distinguer une forme homozygote pour l'instant silencieuse mais qui peut se révéler dans les années ou les décennies qui suivent et une forme hétérozygote complètement bénigne.
Dépistage
Lorsque les deux parents d'un enfant sont porteurs de la maladie, il est indispensable de faire dépister l'enfant avant que les premiers symptômes ne soient visibles ; en général ces symptômes apparaissent à l'adolescence. D'après de nombreux témoignages de malades et de leurs proches les symptômes de la maladie apparaissent à partir de 14 ans.
Notes et références
- ↑ (en) Wilson SAK, « Progressive lenticular degeneration: a familial nervous disease associated with cirrhosis of the liver » Brain, 1912;34;20–509
- ↑ (en) Walshe JM, « Wilson's disease. New oral therapy » Lancet 1956;267;25–26
- 1 2 3 (en) Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky Ml, « Wilson's disease » Lancet, 2007;369:397-408
- ↑ (en) Medici V, Mirante VG, Fassati LR et al. Monotematica AISF 2000 OLT Study Group. « Liver transplantation for Wilson's disease: The burden of neurological and psychiatric disorders » Liver Transpl, 2005;11;1056–1063
- ↑ (en) Kozic D, Svetel M, Petrovic B, Dragasevic N et al. « Imaging of the brain in patients with hepatic form of Wilson's disease » Eur J Neurol. 2003;10;587–592
- ↑ Quemeneur A.S, Ea, H.K & Lioté F (2016) Maladie de Wilson et manifestations ostéoarticulaires. L'actualité rhumatologique 2014, 125.
- ↑ Wendling, D., Vanlemmens, C., Prati, C., Godfrin-Valnet, M., Verhoeven, F., Guillot, X., & Di Martino, V. (2012). Deux cas de spondylarthrite ankylosante et de maladie de Wilson chez le même patient. Seulement une association fortuite ?. Revue du rhumatisme, 79(4), 374-375.
- ↑ (en) Martins da Costa C, Baldwin D, Portmann B, Lolin Y et al. « Value of urinary copper excretion after penicillamine challenge in the diagnosis of Wilson's disease » Hepatology 1992;15;609–615
- ↑ (en) Weiss KH, Thurik F, Gotthardt DN et al. « Efficacy and safety of oral chelators in treatment of patients with Wilson disease » Clin Gastroenterol Hepatol, 2013;11:1028-1035
Sources
Voir aussi
- Intoxication par le cuivre