La cinétique chimique est l'étude de la vitesse des réactions chimiques. Sur le plan disciplinaire, elle fait partie de la chimie physique.
Certaines réactions sont totales et très rapides, voire instantanées, comme les explosions. D'autres sont tellement lentes qu'elles durent plusieurs années (comme la formation de la rouille), voire plusieurs siècles (comme la formation du charbon ou du pétrole). Certaines sont même tellement lentes que les réactifs de départ sont considérés comme stables, par exemple la transformation du diamant en carbone graphite. On parle alors d'états « métastables ».
Importance et intérêt de la cinétique chimique
Importance pratique
Connaître la vitesse des réactions chimiques et être capable de la calculer est de toute première importance dans toutes les applications de la chimie.
Quelques exemples :
- la vitesse de combustion des mélanges utilisés dans les moteurs à explosion, les réacteurs d'avions, les moteurs fusées ;
- la vitesse de prise des colles, des ciments, de polymérisation, de durcissement ;
- la vitesse de dégradation des matériaux, d'oxydation des métaux ;
- la vitesse des réactions chimiques mises en œuvre dans les usines de production de produits chimiques ;
- les vitesses d'action, de dégradation et d'élimination des médicaments (pharmaco-cinétique) ;
- les vitesses de formation, de transformation et d'élimination des polluants dans l'environnement ;
- etc.
La maîtrise de la vitesse de réaction est fondamentale pour un bon usage des substances chimiques et éviter des catastrophes : emballement des réactions, explosions.
Intérêt théorique
La cinétique chimique permet d'établir des lois de vitesse (voir plus loin) qui servent à valider ou infirmer des hypothèses sur les mécanismes réactionnels des réactions chimiques.
Vitesse de réaction
Considérons une réaction dont l'équation chimique s'écrit[1] :

avec , et les coefficients stœchiométriques qui sont généralement des nombres entiers naturels.



Lorsque la réaction progresse, les réactifs disparaissent et les produits se forment en respectant la stœchiométrie de la réaction, c'est-à-dire les proportions indiquées par les coefficients de l'équation chimique. Si pendant l'intervalle il a disparu la quantité de l'espèce :



- il a disparu également la quantité de l'espèce ,
- il est apparu la quantité de l'espèce .




Tous les rapports sont égaux au signe près :


La variable notée est appelée avancement de la réaction : elle quantifie l'évolution de la réaction entre son état initial (avancement nul ) et son état final (avancement maximal)[note 1]. Lorsque la réaction progresse, et . Mais dans un équilibre chimique il se peut que la réaction régresse, auquel cas et . Le tableau suivant résume l'évolution des quantités de réactifs et produits au cours de la réaction.





| Réaction | |||
|---|---|---|---|
| instant | |||
| instant | |||
| variation de quantité | |||













Les variations de quantité de matière pendant la durée pour les espèces sont :
- pour l'espèce : ;
- pour l'espèce : ;
- pour l'espèce : .



La vitesse molaire de réaction est définie par la dérivée de l'avancement de la réaction par rapport au temps[2]:

Elle s'exprime donc en mole par unité de temps. (Nota : on utilise souvent à la place de ).


La vitesse de réaction est une grandeur extensive, donc sa valeur dépend du volume du système. Pour s'affranchir de ce problème, on définit une grandeur intensive, appelée vitesse volumique de réaction. En effet dans le cas fréquent de réactions s'effectuant dans un système de volume constant (c'est en particulier les cas des réactions en solution), la vitesse est définie par rapport à la variation de la concentration. On l'appelle alors vitesse spécifique ou globale volumique de réaction[note 2] :


- , concentration de l'espèce ;
- , son coefficient stœchiométrique.



Dans cette relation (tout comme dans la définition de l'avancement de réaction) est affecté du signe - pour les réactifs et du signe + pour les produits qui se forment. Dans les unités du SI, la vitesse volumique s'exprime en mol m−3 s−1 ou en mol L−1 s−1.
Pour les réactions en phases gazeuse, on peut définir la vitesse par rapport à la dérivée de la pression partielle :

étant la pression partielle du constituant [note 3]. Dans les unités du SI, la vitesse s'exprime alors en Pa s−1[note 4].

Facteurs influençant la vitesse des réactions
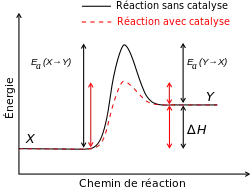
La vitesse des réactions est sous la dépendance de plusieurs facteurs. On peut citer :
- la température,
- la quantité des réactifs présents (en solution c'est la concentration des réactifs qui intervient),
- la pression pour les réactions en phase gazeuse,
- le degré de mélange des réactifs (ségrégation),
- la surface ou l'aire de contact des réactifs dans le cas de systèmes hétérogènes,
- la présence d'un catalyseur qui peut accélérer la réaction ou d'un inhibiteur qui peut la ralentir,
- l'intensité lumineuse (UV ou visible), dans le cas des réactions photochimiques.
Le facteur le plus important est la température, car l'énergie thermique permet en effet dans de nombreux cas de franchir la barrière énergétique qui existe entre le système dans son état initial (mélange de réactifs) et son état final (produits formés).
Un autre paramètre important est l'état de la matière. De ce point de vue, les réactions les plus favorisées sont les réactions qui se déroulent au sein d'une seule phase (liquide : solution, mélange de liquides miscibles ou gazeuse). En effet, dans ces cas, les molécules des réactifs sont dans une même phase et peuvent donc facilement entrer en contact pour réagir.
En solution liquide, les vitesses des réactions rapides sont limitées par la diffusion des réactifs les uns vers les autres.
Dans le cas de systèmes hétérogènes, c'est-à-dire de réactions entre :
- un solide et un gaz,
- un solide et un liquide,
- un solide et un solide,
- un liquide et un gaz,
- deux liquides non miscibles.
La réaction ne peut avoir lieu qu'aux surfaces de séparation des phases (interfaces).
Lorsque les produits sont fractionnés, la réaction est plus rapide, c'est le cas notamment des :
- aérosols (fines gouttelettes de liquide dispersées dans un gaz),
- émulsions (fines gouttelettes d'un liquide dans un autre liquide, dans le cas de liquides non miscibles),
- mélanges de poudres finement broyées (fins grains de solides),
- lysoles (poudre dans un liquide),
- mousses et écumes (bulles dans un liquide).
En effet, dans le cas de la matière fractionnée, la surface de contact entre les réactants est importante, donc les possibilités de réaction nombreuses. Pour les solides, on quantifie ceci par la surface spécifique, qui est la surface libre par unité de masse ; une poudre, un solide poreux ou une mousse (filaments imbriqués) ont une grande surface spécifique. Lorsque les réactants ne sont pas dans le même état (par exemple solide-gaz, solide-liquide, liquide-gaz), on parle de réaction hétérogène.
Cas de l'activation thermique
La température est le facteur ayant la plus grande influence sur la vitesse des réactions chimiques. Dans un très grand nombre de cas la vitesse de la réaction augmente d'un facteur 2 à 3 lorsque la température augmente de 10 degrés. La dépendance de la vitesse de réaction en fonction de la température a fait l'objet d'études expérimentales intensives à la fin du XIXe siècle. Plusieurs équations empiriques ont été proposées, dont les lois d'Arrhenius et d'Eyring-Polanyi. Ces deux lois indiquent que la vitesse de réaction augmente en fonction de la température. La loi d'Arrhenius, qui a été vérifiée pour un très grand nombre de réactions, est formulée ainsi :

où est appelé l'énergie d'activation (dans sa version décrite ici, la loi d'Arrhenius suppose que l'énergie d'activation est indépendante de la température), la constante des gaz parfaits, la température en kelvin, la constante de vitesse de la réaction et un facteur pré-exponentiel dépendant de la réaction. La loi d'Arrhenius est compatible avec la théorie des collisions qui permet de jeter un pont entre la représentation microscopique (à l'échelle des molécules) de la réaction chimique et l'observation macroscopique de sa vitesse.




La Loi d'Eyring est donnée par l'expression suivante :

avec l'enthalpie libre d'activation , l'enthalpie d'activation, l'entropie d'activation, la constante de Boltzmann et la constante de Planck. L'équation d'Eyring repose sur la théorie de l'état de transition (aussi appelée théorie du complexe activé).






Toutes les réactions ne suivent pas les lois d'Arrhenius et d'Eyring. C'est en particulier le cas des réactions explosives, des réactions catalysées par les enzymes ou ayant d'autres modes d'activation que l'activation purement thermique (activation par rayonnement, par électrochimie ou par micro-ondes).
Loi de vitesse et ordre de réaction
On appelle loi de vitesse ou équation de vitesse la relation qui existe entre la vitesse et tous les facteurs qui ont une influence sur celle-ci. Dans le cas général, la loi de vitesse ne peut pas être déduite directement de l'équation chimique de la réaction. Elle doit être établie expérimentalement à partir de séries d'expériences judicieusement conçues, en faisant varier les différents facteurs.
Parmi ces facteurs, on distingue d'une part les facteurs physiques (et en particulier la température) et, d'autre part, les quantités (ou les concentrations) des substances présentes dans le milieu réactionnel (et en particulier les réactifs).
La loi de vitesse peut se mettre sous la forme d'un produit de deux fonctions :

Le premier facteur est le coefficient de vitesse (ou constante de vitesse) qui dépend notamment de la température, le second est une fonction des quantités (ou des concentrations) des substances présentes dans le milieu réactionnel.

Dans de nombreux cas, la fonction peut se mettre sous la forme d'un monôme :


On dit alors que la réaction admet un ordre et l'on appelle ordre de réaction la somme des exposants : . L'exposant est l'ordre partiel de la réaction par rapport à l'espèce .



Les ordres partiels ne sont pas nécessairement égaux aux coefficients stœchiométriques, ils ne sont même pas nécessairement entiers ni positifs, ce peut être des nombres fractionnaires et même des nombres réels. Si la loi de vitesse ne peut pas se mettre sous la forme d'une loi de puissance avec les concentrations comme facteurs, on dit que la réaction n'admet pas d'ordre. C'est la situation classique en cinétique enzymatique.
Les réactions élémentaires
Une réaction est dite élémentaire si les réactifs réagissent simultanément en un même point pour donner directement les produits sans former d'espèces intermédiaires[3]. La vitesse de réaction dépend donc de la probabilité de rencontre des réactifs donc de la fréquence des chocs. Cette fréquence est proportionnelle à leur concentration. Par ailleurs, plus la température est élevée, plus les rencontres sont probables (agitation thermique) et plus l'énergie cinétique des réactifs au moment du choc est élevée ce qui permet de franchir la barrière d'activation, donc la température a également un rôle important.
On appelle molécularité le nombre d'entités (molécules, ions) qui entrent simultanément en contact lors d'une réaction élémentaire. Les réactions élémentaires sont dites monomoléculaires, bimoléculaire ou trimoléculaire selon la valeur de leur molécularité (respectivement 1, 2 ou 3). Comme la probabilité que plus de 3 entités se trouvent simultanément en un même point est quasiment nulle, on considère qu'il n'existe pas de réaction élémentaire de molécularité supérieure à 3 (la probabilité de chocs triples étant elle-même très faible, l'existence de réactions élémentaires trimoléculaires est discutée).
Pour une réaction élémentaire, son ordre est égal à sa molécularité (loi de van 't Hoff).
Par exemple, pour une réaction élémentaire bimoléculaire :
la loi de vitesse prendrait la forme :
![v=-{\frac {{\mathrm d}[A]}{{\mathrm d}t}}=k\cdot [{{\rm {A}}}]\cdot [{{\rm {B}}}]](https://img.franco.wiki/i/cb689a6dae84fd11624545267923f9796a7c6d15.svg)
d'ordre global 2 (ordre partiel 1 par rapport à chaque réactif). Évidemment, il est également possible de définir la vitesse sur les constituants B ou C. Ici k est le coefficient de vitesse dépendant notamment de la température et éventuellement d'autres facteurs physiques.
Les réactions composées
Au cours d'une transformation chimique, plusieurs réactions élémentaires peuvent avoir lieu simultanément ou successivement. On appelle réactions composées (ou complexes) des ensembles d'étapes élémentaires, combinées par exemple selon un des trois schémas simples suivants: série, opposé ou parallèle. Les mécanismes en chaîne font une classe plus compliqué[4].
Réactions en série
Le ou les produit(s) d'une réaction peuvent eux-mêmes être les réactifs d'une autre réaction. Cela constitue un système de réactions dites en série ou par stades ou en séquence ouverte. Les réactions se produisent dans un ordre donné, toujours le même, des réactifs aux produits.
Pour une réaction possédant plusieurs étapes, la vitesse globale de formation du produit final est déterminée par la vitesse de l'étape la plus lente, dite l'étape cinétiquement déterminante.
Réactions opposées
Certaines réactions peuvent se produire dans les deux sens. On les appelle des réactions opposées. Dans ce cas, le système évolue vers un état d'équilibre dynamique.
Du point de vue macroscopique, la réaction semble terminée puisque la composition du système ne varie plus. Le taux d'avancement α est alors compris entre 0 et 1 et la réaction n'est pas totale. Mais du point de vue microscopique les réactions directes et inverses continuent de se produire avec des vitesses égales.
Réactions parallèles ou simultanées
On appelle réactions parallèles des systèmes de réactions distinctes qui ont les mêmes réactifs mais des produits différents. Par exemple la nitration du phénol forme simultanément les trois isomères ortho, para et méta du mono-nitrophénol[5]. La vitesse de transformation des réactifs est alors la somme des vitesses de chacune des réactions.
Cinétique des réactions complexes
Une réaction complexe peut se décomposer en plusieurs réactions élémentaires. On appelle mécanisme réactionnel l'ensemble des réactions élémentaires mises en jeu. Ce mécanisme ne peut pas être observé directement, il constitue donc une hypothèse qui doit être confrontée à l'ensemble des observations que l'on peut faire sur le déroulement de la réaction. La loi de vitesse établie expérimentalement dépend du mécanisme par lequel la réaction a lieu. La cinétique est donc une approche incontournable pour vérifier la validité des mécanismes réactionnels.
Cependant il peut arriver (mais cela est assez rare pour une réaction complexe) qu'on retrouve une loi de vitesse qui admette un ordre :

comme cela a été précisé plus haut les ordres partiels ne sont pas nécessairement égaux aux coefficients stœchiométriques, ils ne sont même pas nécessairement entiers ni positifs.
Mécanismes en chaîne
Aussi appelé séquence fermée. Ici, un intermédiaire réactionnel (ou centre actif) est régénéré au bout d'un certain nombre d'étapes qui se reproduisent de manière cyclique. Ainsi le centre actif pourra faire réagir un grand nombre de réactifs. Le centre actif met en jeu des intermédiaires réactionnels dont les concentrations stationnaires pourront être calculables par le principe de Bodenstein.
Les étapes types des réactions en chaîne sont :
- l'initiation ou la formation des radicaux libres ou autres molécules instables comme porteurs de chaîne ;
- la propagation de la chaîne catalysée par les porteurs de chaîne ;
- la rupture ou la terminaison de la chaîne par la recombinaison des radicaux porteurs de chaîne.
Il arrive que la chaîne de réaction soit ramifiée (i.e. plusieurs centres actifs, comme dans les oxydations) et cela entraîne souvent des réactions explosives dues aux multiplications des centres réactifs et à l'énergie dégagée lors des terminaisons.
La longueur de chaîne est définie comme le nombre de cycles que peut effectuer en moyenne un radical à partir de sa formation avant d'être détruit :


- , vitesse de propagation de la chaîne ou vitesse de consommation du réactif ;
- , vitesse d'initiation ou vitesse de formation du radical par les méthodes d'initiation.


Exemple de mécanisme en chaîne : H2 + Br2 → 2HBr
La réaction entre le dihydrogène gazeux et le dibrome gazeux est un excellent exemple de réaction complexe.
La loi de vitesse n'a pas la forme simple d'un monôme et alors on dit qu'elle n'admet pas d'ordre. Elle possède plutôt une forme plus complexe avec un dénominateur à deux termes[6] :
![{\frac {{\mathrm d}[HBr]}{{\mathrm d}t}}={\frac {k[H_{2}][Br_{2}]^{{3/2}}}{[Br_{2}]+k'[HBr]}}](https://img.franco.wiki/i/24b1ee521a4c94b3c4c3c24184c866a68c103dbd.svg)
Cette loi s'explique par un mécanisme qui fait intervenir deux intermédiaires radicalaires H. et Br. dans une réaction en chaîne. Voir Réaction chimique en chaîne#Exemple.
Bibliographie
- Paul Arnaud, Cours de chimie physique, Dunod (1990).
- Marc Laffitte et Françoise Rouquérol, La Réaction chimique, tome 2: aspects thermodynamiques (suite) et cinétiques, Masson, Paris (1991), 306 p. (ISBN 2-225-82156-9)
Références
- ↑ « cinétique »
- ↑ Union internationale de chimie pure et appliquée. Quantities, Units and Symbols in Physical Chemistry [« Green Book »], Oxford, Blackwell Science, , 2e éd. (ISBN 0-632-03583-8, lire en ligne), p. 55.
Traduction française: Manuel des symboles et de terminologie des grandeurs et des unités physicochimiques. Société Chimique de France. Supplément à L'actualité chimique, 1982,No 9. - ↑ (en) « elementary reaction », IUPAC, Compendium of Chemical Terminology [« Gold Book »], Oxford, Blackwell Scientific Publications, 1997, version corrigée en ligne : (2019-), 2e éd. (ISBN 0-9678550-9-8).
- ↑ (en) « composite mechanism », IUPAC, Compendium of Chemical Terminology [« Gold Book »], Oxford, Blackwell Scientific Publications, 1997, version corrigée en ligne : (2019-), 2e éd. (ISBN 0-9678550-9-8).
- ↑ M.Prettre et B.Claudel, Éléments de cinétique chimique (Gordon et Breach 1969), p. 75-76.
- ↑ P. Atkins et J. de Paula, Chimie physique (3e éd. française, de Boeck 2008) p. 831.
Notes
- ↑ Dans l'équation chimique, les coefficients stœchiométriques sont positifs, tandis que dans la définition de l'avancement de réaction, on affecte le signe - aux coefficients des réactifs qui disparaissent, et le signe + aux coefficients des produits qui se forment.
- ↑ Lorsqu'il n'y a pas risque de confusion, on omet souvent le qualificatif « volumique ».
- ↑ La pression partielle d'un constituant d'un mélange gazeux est égale à la pression, qui serait celle de ce constituant s'il était seul à occuper tout le volume du système considéré. Elle peut se calculer directement en utilisant l'équation état du gaz ou en multipliant la fraction molaire par la pression totale.
- ↑ On utilise souvent à la place de .

Voir aussi
Articles connexes
- Catalyseur
- Cinétique électrochimique
- Énergie cinétique
- Énergie d'activation
- Loi d'Arrhenius
- Mécanisme réactionnel
- Ordre de réaction
- Saut de température (méthode pour réactions très rapides)
- Théorie des collisions
- Théorie du complexe activé