|
Équilibre chimique
| |
| Notions de base | |
|---|---|
| Réaction chimique | Équation bilan : espèce chimique. |
| Stœchiométrie | Coefficients stœchiométriques :
|
| Avancement de réaction | quantité du constituant . |
| Affinité chimique | potentiel chimique. |
| Deuxième principe de la thermodynamique | Condition d'évolution spontanée : à l'équilibre. |
| Réaction à pression et température constantes | |
| Enthalpie libre | Enthalpie libre de réaction : |
| Constante d'équilibre | État standard : |
| Quotient de réaction | activité chimique. |
| Progression de la réaction | Déplacement des réactifs vers les produits : , , |
| Régression de la réaction | Déplacement des produits vers les réactifs : , , |
| Condition d'équilibre | Loi d'action de masse : , |
| Condition de stabilité | strictement. |
| Déplacement d'un équilibre chimique | |
| Principe de modération de Le Chatelier | « Lorsque les modifications extérieures apportées à un système physico-chimique en équilibre provoquent une évolution vers un nouvel état d'équilibre, l'évolution s'oppose aux perturbations qui l'ont engendrée et en modère l'effet. » |
| Ce principe n'est vrai que pour les paramètres intensifs conditionnant l'équilibre. Il existe des contre-exemples pour les paramètres extensifs. | |
| Augmentation de la température | À pression ou volume constant, déplacement dans le sens endothermique (loi de van 't Hoff). |
| Augmentation de la pression | À température constante, déplacement dans le sens d'une diminution du volume, de la quantité totale de constituants (loi de Le Chatelier). |
| Augmentation du volume | À température constante, déplacement dans le sens d'une augmentation de la pression, de la quantité totale de constituants. |
| Ajout d'un réactif ou d'un produit | À pression et température constantes, pour les solutions idéales, déplacement dans le sens qui diminue la fraction molaire du constituant. |
| À volume et température constants, pour les mélanges de gaz parfaits, déplacement dans le sens qui diminue la quantité du constituant. | |
| Ajout d'un inerte | À pression et température constantes, pour les solutions idéales, déplacement dans le sens d'une augmentation de la quantité totale de constituants. |
| À volume et température constants, pour les mélanges de gaz parfaits, absence de conséquence. | |
| Emploi d'un catalyseur | L'équilibre est atteint plus vite, mais il n'est pas déplacé. |
| Équilibres chimiques simultanés | |
| Couplage chimique | Une réaction endergonique qui, seule, ne pourrait être spontanée, peut être provoquée par couplage avec une réaction exergonique simultanée. |


























Un équilibre chimique est le résultat de deux réactions chimiques simultanées dont les effets s'annulent mutuellement.
Une réaction telle que la combustion du propane avec l'oxygène, qui s'arrête lorsque l'un des réactifs est totalement épuisé, est qualifiée de réaction totale, complète ou irréversible. À contrario, une réaction comme l'estérification, aboutissant à un mélange stable dans le temps de réactifs et de produits, sans disparition totale de l'une des espèces chimiques, est qualifiée de réaction partielle, incomplète, réversible ou inversible[alpha 1] : ce type de réaction aboutit à un équilibre chimique. Au cours d'un processus de transformation chimique deux réactions peuvent s'opposer, l'une consommant des réactifs, l'autre consommant les produits de la première réaction pour recréer les réactifs initiaux. Une réaction est totale lorsqu'elle l'emporte sur sa réaction antagoniste. Un équilibre chimique apparaît lorsque la première réaction consomme les réactifs aussi vite que la seconde les recrée.
Une modification des conditions opératoires d'un équilibre chimique (modification de la pression ou de la température, ajout ou extraction de l'un des constituants du mélange réactionnel, etc.) peut favoriser l'une ou l'autre réaction. Ceci implique un déplacement de l'équilibre, c'est-à-dire l'obtention d'un nouvel état d'équilibre à une composition différente de celle de l'équilibre initial, mais contenant toujours les mêmes espèces. Un retour aux conditions opératoires initiales induit un retour à l'équilibre initial. Dans certains cas, la modification des conditions opératoires peut conduire à une rupture d'équilibre, c'est-à-dire l'obtention d'une réaction totale avec disparition de l'une des espèces.
Une réaction chimique se déplace spontanément dans le sens dicté par le deuxième principe de la thermodynamique. Une réaction qui diminue l'entropie ne peut par conséquent se produire seule, mais elle peut être provoquée. Dans un milieu réactionnel qui augmente globalement l'entropie, en présence d'autres réactions, un couplage chimique peut permettre d'obtenir une réaction qui serait impossible seule.
Historique
La notion d'équilibre chimique est évoquée pour la première fois en 1803 par le chimiste français Claude-Louis Berthollet, à la suite de ses observations sur les rives du lac Natron lors de la campagne d'Égypte. Avant lui les réactions chimiques étaient supposées être toujours totales. Berthollet est le premier à observer et étudier un mélange de réactifs et de produits (en l'occurrence un mélange de carbonate et chlorure de calcium et de sodium) dont l'équilibre se déplace tantôt dans un sens tantôt dans l'autre, selon les conditions de température et de composition. Il en déduit que les réactions chimiques sont régies par des forces opposées, mais ne peut préciser la nature de celles-ci[1],[2],[3].
Il faut attendre la deuxième moitié du XIXe siècle pour que des progrès significatifs soient faits dans la compréhension des réactions et équilibres chimiques. En 1858 le physicien allemand Gustav Kirchhoff énonce sa relation liant la variation de la chaleur d'une réaction chimique à la différence des capacités calorifiques des produits et des réactifs. En 1865 les chimistes norvégiens Cato Guldberg et Peter Waage, en s'inspirant des propositions de Berthollet, montrent expérimentalement qu'il existe une relation entre les concentrations des espèces présentes à l'équilibre en solution. La constante d'équilibre qu'ils définissent est appelée « constante de Guldberg et Waage » ou constante de la loi d'action de masse. Un premier formalisme mathématique des équilibres chimiques est développé par le chimiste néerlandais Jacobus van 't Hoff. La relation portant son nom donne la variation de la constante d'équilibre en fonction de la chaleur de réaction[3].


En 1884 le chimiste français Henry Le Chatelier, toujours sur base d'expérimentations, énonce son « principe de modération », dit « principe de Le Chatelier », selon lequel un équilibre s'oppose aux changements extérieurs qui tentent de le modifier[3]. Il existe cependant des cas dans lesquels ce principe n'est pas vérifié, notamment pour l'ajout ou l'extraction d'une espèce du mélange réactionnel[4],[5]. Ce principe est également appelé « principe de Le Chatelier-Braun », du nom du physicien allemand Ferdinand Braun qui en donna en 1887 une première démonstration mathématique[6].
Les bases théoriques de la chimie physique sont posées par le physicien américain Willard Gibbs entre 1875 et 1878. Gibbs introduit les fonctions enthalpie libre et potentiel chimique [3]. En 1922 le physicien belge Théophile de Donder définit l'avancement de réaction et l'affinité chimique . Il relie ainsi l'évolution des réactions chimiques au deuxième principe de la thermodynamique et donne la condition d'évolution spontanée de toute réaction chimique[7],[8]. En 1923 le physicien américain Gilbert Lewis introduit l'activité chimique [9]. Ce formalisme mathématique permet en 1950 aux physiciens belges Ilya Prigogine et Raymond Defay[alpha 2] de démontrer rigoureusement les relations et principes formulés par leurs prédécesseurs[10].





Notions de base
Principe de Berthelot, principe de Matignon
Une réaction chimique est favorisée ou défavorisée par divers facteurs tels que la pression, la température, l'énergie apportée au mélange réactionnel. L'étude des réactions chimiques et des équilibres a conduit, dans la seconde moitié du XIXe siècle, à l'énoncé des deux principes empiriques suivants[11],[12],[13] :
« Principe de Berthelot - Facteur énergie.
Un système laissé à lui-même tend à évoluer de manière à libérer le plus de chaleur. »
Autrement dit, une réaction est favorisée si elle est exothermique, elle est défavorisée si elle est endothermique. Les réactions exothermiques ayant une très forte chaleur de réaction (enthalpie standard de réaction ), comme les réactions de combustion, sont souvent totales.

« Principe de Matignon - Facteur désordre.
Une réaction chimique évolue dans le sens de l'augmentation du nombre de ses molécules. »
Les réactions augmentant la quantité de matière dans le mélange réactionnel sont favorisées : la réaction de combustion d'un alcane avec l'oxygène est favorisée, car le nombre de moles de produits est supérieur à celui des réactifs ; en outre elle est exothermique, elle est donc doublement favorisée ce qui la rend totale. Le principe de Matignon est en accord avec le deuxième principe de la thermodynamique : les réactions dont l'entropie standard de réaction est positive sont favorisées, or une augmentation du nombre de moles d'un système thermodynamique à la suite d'une réaction correspond à une augmentation de l'entropie. Les réactions produisant un gaz à partir d'un solide ou d'un liquide sont donc également favorisées, car l'entropie d'un gaz est plus élevée que celle d'une phase condensée[alpha 3].

Lorsque ces deux principes, qui en eux-mêmes n'ont qu'un intérêt historique, entrent en concurrence au sein d'une même réaction, il apparaît un équilibre. Leur combinaison, qui fut effectuée par Gibbs et Duhem[alpha 4], conduit à l'étude de l'enthalpie libre standard de réaction , définie par :


avec la température à laquelle est effectuée la réaction. Ainsi[11],[12],[13] :

- lorsque et sont de signes opposés, les deux principes sont simultanément favorables ou défavorables :
- si et , soit , selon les deux principes tous les facteurs sont défavorables : la réaction est impossible ;
- si et , soit , selon les deux principes tous les facteurs sont favorables : la réaction est totale ;
- lorsque et sont de même signe, les deux principes s'opposent, peut être positive ou négative : la réaction conduit à un équilibre d'autant plus déplacé que est éloignée de 0 :
- si la réaction est peu favorisée, l'équilibre contiendra majoritairement des réactifs, peu consommés ;
- si la réaction est favorisée, l'équilibre contiendra majoritairement des produits, les réactifs étant en grande partie consommés.








Le tableau suivant récapitule l'influence des deux facteurs sur la réaction[11],[12].
| Facteurs influant sur la réaction | Principe de Matignon | ||
|---|---|---|---|
| Augmentation du désordre favorable | Diminution du désordre défavorable | ||
| Principe de Berthelot | Réaction exothermique favorable | Réaction totale | Équilibre exothermique |
| Réaction endothermique défavorable | Équilibre endothermique | Réaction impossible | |
L'eau oxygénée H2O2 liquide se décompose en eau H2O liquide et en oxygène O2 gazeux selon :

Cette réaction est exothermique ( = −106,7 kJ/mol = −25,5 kcal/mol) et est donc favorisée selon le principe de Berthelot. Elle augmente le nombre de ses molécules, une mole d'eau oxygénée produisant une mole d'eau et une demie mole d'oxygène, soit une mole et demie de produits, elle est donc favorisée selon le principe de Matignon. De plus elle produit un gaz à partir d'un liquide, ce qui la favorise également. Il s'agit donc d'une réaction totale.
La réaction inverse :

est endothermique, diminue le nombre de ses constituants et produit un liquide à partir d'un gaz. Elle est triplement défavorisée, donc impossible.
La réaction de synthèse en phase gaz du trioxyde de soufre SO3 à partir du dioxyde de soufre SO2 et de l'oxygène O2 s'écrit :

La chaleur de réaction est de = −99,6 kJ/mol = −23,8 kcal/mol : la réaction est exothermique, elle est donc favorisée selon le principe de Berthelot. Cependant le nombre de constituants décroît selon cette réaction : pour une mole et demie de réactifs il ne reste qu'une mole de produit, le principe de Matignon énonce donc que cette réaction n'est pas favorisée.
Si l'on écrit la réaction dans le sens inverse, les facteurs sont également inversés :

Cette réaction est endothermique et donc n'est pas favorisée selon le principe de Berthelot, mais elle accroît le désordre et est donc favorisée selon le principe de Matignon.
Les deux principes s'opposant, il s'ensuit un équilibre exothermique ( = −99,6 kJ/mol) noté :

Noté dans l'autre sens, l'équilibre est endothermique ( = 99,6 kJ/mol) :

En toute rigueur cependant, une réaction telle que et n'est pas impossible. Elle ne peut pas être spontanée, c'est-à-dire avoir lieu seule, car elle diminue l'entropie, ce qui est contraire au deuxième principe de la thermodynamique. Néanmoins elle peut être provoquée : cette réaction pourrait par exemple avoir lieu par couplage chimique avec une autre réaction, de telle sorte que l'entropie globale créée par ces deux réactions soit positive, en accord avec le deuxième principe[14] (voir le chapitre Équilibres chimiques simultanés).
D'autre part, si l'un des facteurs et est significativement plus important que l'autre, une réaction à priori équilibrée peut être en réalité totale ou impossible. C'est le cas de la combustion du monoxyde de carbone CO en dioxyde de carbone CO2 en présence d'oxygène O2 :

Cette réaction est exothermique et diminue le nombre de ses molécules : elle est donc favorisée selon Berthelot et défavorisée selon Matignon, elle devrait être équilibrée. Cependant sa très forte exothermicité ( = −67,6 kcal/mol = −282,8 kJ/mol) la rend totale jusqu'à environ 1 000 °C[12].
Enfin, si les deux facteurs et sont peu marqués, une réaction à priori totale ou impossible peut être en réalité équilibrée. C'est le cas de la synthèse de l'iodure d'hydrogène HI en présence d'iode I2 et d'hydrogène H2 :

Cette réaction est faiblement exothermique ( = −9,4 kJ/mol) et ne modifie pas le nombre de ses molécules : elle devrait être totale selon Berthelot, mais est en fait équilibrée[12].
L'enthalpie libre standard de réaction ne tient compte que de la stœchiométrie et de la température, le mélange réactionnel étant considéré dans un état standard généralement différent des conditions réelles de réaction. Une étude plus fine des réactions chimiques passe par l'utilisation de l'enthalpie libre de réaction , qui, en plus de la température et de la stœchiométrie, prend en compte la pression et la composition du mélange réactionnel, et les activités chimiques des constituants[13].

Réaction directe et réaction inverse, cinétique des réactions
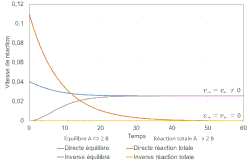
Dans une réaction totale, la vitesse de la réaction directe finit par s'annuler ; la réaction inverse est inexistante. Dans un équilibre chimique les vitesses finales des réactions directe et inverse s'égalisent et sont non nulles.
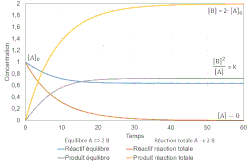
Dans une réaction totale, un réactif limitant, c'est-à-dire un réactif introduit en défaut par rapport à la stœchiométrie, disparaît totalement. Dans un équilibre, le mélange réactionnel final contient toutes les espèces de réactifs et de produits en quantités non nulles.
Un équilibre chimique implique deux réactions simultanées :
- une réaction dans le sens réactifs → produits appelée réaction directe, il s'agit généralement de la réaction intéressante pour la synthèse d'une espèce chimique donnée ;
- une réaction dans le sens produits → réactifs appelée réaction inverse, qui s'oppose à la réaction directe.
Dans un équilibre, l'une des deux réactions obéit au principe de Berthelot et l'autre au principe de Matignon : si l'une est exothermique, l'autre augmente le désordre du système réactionnel ; en conséquence, puisque les deux réactions ont des effets inverses, la première enfreint le principe de Matignon en diminuant le désordre, et la deuxième enfreint le principe de Berthelot en étant endothermique.
Lorsqu'une réaction est équilibrée, cela signifie que la vitesse de la réaction directe est égale à la vitesse de la réaction inverse. L'état d'équilibre obtenu dans ce cas peut être qualifié d'équilibre dynamique ou stationnaire : les réactions ont toujours lieu, mais globalement leurs effets s'annulent ; par exemple la réaction directe consomme les réactifs aussi vite que la réaction inverse les recrée, d'où une composition du mélange réactionnel stable dans le temps ; ou encore la réaction endothermique absorbe totalement la chaleur dégagée par la réaction exothermique, d'où un bilan énergétique globalement nul.
Dans le cas d'un équilibre impliquant deux réactifs et et deux produits et , on note l'équilibre sous la forme :





où , , et sont des espèces chimiques, , , et les coefficients stœchiométriques respectifs.




Dans le cas de réactions élémentaires, c'est-à-dire s'effectuant en une seule étape, les vitesses de réaction dépendent des concentrations , , et des espèces en présence et de et les constantes respectives des vitesses des réactions directe et inverse (qui suivent la loi d'Arrhenius) selon les expressions :
![{\displaystyle {\rm {\left[A\right]}}}](https://img.franco.wiki/i/1a9d770ceca6b98c80bada48786413f022f7ace4.svg)
![{\displaystyle {\rm {\left[B\right]}}}](https://img.franco.wiki/i/07e89ce8f705c242a799cfb9853e1d2e4988d45d.svg)
![{\displaystyle {\rm {\left[C\right]}}}](https://img.franco.wiki/i/31c044eb69d03082ed8f885e8ea2f452bd592fb1.svg)
![{\displaystyle {\rm {\left[D\right]}}}](https://img.franco.wiki/i/e075477e9777230ecdfea90bb54f5f00c4ef0dd6.svg)


- la vitesse de la réaction directe ;
- la vitesse de la réaction inverse.
![{\displaystyle v_{\rightarrow }=k_{\rightarrow }\left[{\rm {A}}\right]^{\nu _{\rm {A}}}\left[{\rm {B}}\right]^{\nu _{\rm {B}}}}](https://img.franco.wiki/i/724c473af9094391f6fea65a65d4385546a9a731.svg)
![{\displaystyle v_{\leftarrow }=k_{\leftarrow }\left[{\rm {C}}\right]^{\nu _{\rm {C}}}\left[{\rm {D}}\right]^{\nu _{\rm {D}}}}](https://img.franco.wiki/i/b17e2b29b3708f4ba015b5e36f1ee36794b2e77d.svg)
Dans une réaction totale, la réaction inverse est inexistante, ou du moins a-t-elle une vitesse négligeable devant la vitesse de la réaction directe : . À la fin d'une réaction totale, c'est-à-dire lorsque les concentrations des réactifs et produits ne varient plus, les réactifs ont totalement disparu s'ils ont été introduits à la stœchiométrie, sinon c'est le réactif introduit en défaut, ou réactif limitant, qui a totalement disparu.

Dans un équilibre chimique, la vitesse de déplacement de l'équilibre est égale à la différence entre la vitesse de la réaction directe et la vitesse de la réaction inverse : . Les deux vitesses de réaction finissent par s'égaliser sans s'annuler : (c'est la vitesse de l'équilibre qui s'annule ). Ceci entraîne, à l'équilibre, la relation suivante, appelée loi d'action de masse :



![{\displaystyle {k_{\rightarrow } \over k_{\leftarrow }}={\left[{\rm {C}}\right]^{\nu _{\rm {C}}}\left[{\rm {D}}\right]^{\nu _{\rm {D}}} \over \left[{\rm {A}}\right]^{\nu _{\rm {A}}}\left[{\rm {B}}\right]^{\nu _{\rm {B}}}}=K}](https://img.franco.wiki/i/37e69a58411323fda1f3ee4def8220fbd83721f9.svg)
Cette relation lie les constantes des vitesses des deux réactions aux concentrations des réactifs et des produits à l'équilibre. Le rapport ainsi obtenu est appelé constante d'équilibre. Cette relation a été établie empiriquement par Guldberg et Waage. Nous verrons par la suite comment établir rigoureusement cette relation selon les principes de la thermodynamique. Dans un équilibre chimique aucune espèce, que ce soit un réactif ou un produit, ne disparaît totalement.
Les expressions correctes des vitesses de réaction et de la constante d'équilibre doivent faire intervenir les activités chimiques des constituants, et non leur concentration ou leur pression molaire partielle souvent utilisées par approximation.
Stœchiométrie, avancement de réaction
Considérons un équilibre chimique réalisé à température et pression constantes dont l'équation bilan est la suivante :

Les constituants du membre de gauche sont les réactifs, au nombre de ; les constituants du membre de droite sont les produits, au nombre de :




- réactif ;
- produit.
![{\displaystyle i\in [1,\cdots ,R]}](https://img.franco.wiki/i/f11e1deb0b9bf8122e4bb5b8cc3207b62d0850fe.svg)
![{\displaystyle j\in [1,\cdots ,P]}](https://img.franco.wiki/i/26e1e9368a77cb6d36f2f684158b9b2e844944d4.svg)
Les coefficients et sont les coefficients de stœchiométrie de l'équation bilan. Considérons une petite progression de la réaction à l'instant et appelons les variations élémentaires du nombre de moles de chaque constituant à ce stade de la réaction. La réaction progresse en respectant la stœchiométrie de l'équation bilan, aussi si la quantité du réactif varie de :






- la quantité de tout réactif varie de : ;
- la quantité de tout produit varie de : ;


ce qui implique que tous les rapports sont égaux au signe près :


Le paramètre est appelé avancement de réaction de la réaction, il est donc défini sur l'un quelconque des constituants et il existe une relation entre et chaque constituant. La connaissance de permet de connaître la composition du système : c'est une variable de composition extensive (si l'on double la quantité de chacun des réactifs et produits, l'avancement de réaction double également). Au début de la réaction, au moment de l'introduction des constituants dans le réacteur, par définition l'avancement est nul : . Puis la valeur de l'avancement évolue selon le sens de déplacement de la réaction.

Lorsque la réaction progresse :
- les réactifs disparaissent : ;
- les produits apparaissent : ;
- l'avancement de réaction augmente : ;
- la réaction directe l'emporte sur la réaction inverse : et ;
- l'équilibre se déplace de la gauche (réactifs) vers la droite (produits).






À l'inverse, lorsque la réaction régresse :
- les réactifs apparaissent : ;
- les produits disparaissent : ;
- l'avancement de réaction diminue : ;
- la réaction directe est dominée par la réaction inverse : et ;
- l'équilibre se déplace de la droite (produits) vers la gauche (réactifs).




L'avancement de réaction peut donc indifféremment augmenter ou diminuer. Puisque l'avancement est nul au début de la réaction, il peut prendre aussi bien une valeur positive (la réaction a progressé par rapport à l'état initial) qu'une valeur négative (la réaction a régressé par rapport à l'état initial).


La convention stœchiométrique communément admise afin de simplifier les expressions dans les démonstrations qui suivent est de représenter l'équilibre sous la forme[15] :
Cette équation (2) implique les mêmes constituants et que l'expression de l'équilibre (1), mais ils sont cette fois notés , au nombre de , en attribuant une valeur négative aux coefficients stœchiométriques des réactifs, et positive à ceux des produits :

- pour un réactif ;
- pour un produit ;
- pour un inerte.
Ceci permet d'écrire indifféremment pour un réactif ou un produit :
| Avancement de réaction : pour tout |

![{\displaystyle k\in [1,\cdots ,N]}](https://img.franco.wiki/i/136b4a52f5c0353b131ec00d25a812da468c7464.svg)
L'urée peut être synthétisée selon la réaction :

La réaction est réécrite en :

Sous cette forme, les coefficients de stœchiométrie valent :
- pour les réactifs :
- ammoniac : ;
- dioxyde de carbone : ;
- pour les produits :







La variation de l'avancement de réaction vaut alors :

Si la réaction progresse dans le sens de la production de l'urée (). Si la réaction régresse dans le sens de la destruction de l'urée en ammoniac ().


À tout instant au cours de la réaction, l'intégration de ces relations permet de calculer la quantité d'un réactif ou d'un produit quelconque, selon :


avec :
- la quantité de constituant à l'instant initial ;
- la quantité de constituant à l'instant ;
- l'avancement de réaction à l'instant ; rappelons qu'à l'instant initial, par définition, .



Pour un inerte, constituant n'intervenant pas dans la réaction, , la quantité ne varie pas au cours de la réaction : on a à tout instant.

La vitesse de déplacement de l'équilibre est égale à la dérivée de l'avancement de réaction par rapport au temps [16],[17] :

La vitesse de réaction peut prendre aussi bien une valeur positive lorsque la réaction progresse (l'avancement de réaction croît) que négative lorsque la réaction régresse (l'avancement de réaction décroît) ; à l'équilibre . Pour tout constituant (réactif, produit ou inerte), la vitesse d'apparition est égale à[16],[17] :

Comme la vitesse de réaction, la vitesse d'apparition d'un constituant peut être positive (le constituant apparait) ou négative (le constituant disparait).
Affinité chimique, condition d'évolution spontanée et condition d'équilibre
L'affinité chimique est une fonction d'état définie à partir des potentiels chimiques [18] :
| Affinité chimique : |
Considérant la relation pour tout constituant :

on a la relation :

L'équilibre du dioxyde et du trioxyde de soufre en présence d'oxygène s'écrit :

On a l'affinité chimique et la variation de l'avancement de réaction :


et la relation :

Le deuxième principe de la thermodynamique implique que l'évolution spontanée d'une réaction chimique ne peut se faire que si[19] :
| Condition d'évolution spontanée : |
et ne peuvent donc être que de même signe[19] :

- une progression est associée à un signe positif : et , la réaction progresse ;
- une régression est associée à un signe négatif : et , la réaction régresse.


Un système fermé est à l'équilibre lorsque les variables intensives qui le décrivent (température, pression et potentiels chimiques des réactifs et des produits) sont homogènes dans tout le système et restent constantes au cours du temps. L'équilibre est atteint lorsque l'affinité est nulle[19] :
| Condition d'équilibre : |
En termes de potentiel thermodynamique, ces relations dépendent des conditions opératoires maintenues constantes en cours de réaction :
- pour une réaction chimique effectuée à et constantes : ,
- la fonction enthalpie libre ne peut que décroître[alpha 5] :
Condition d'évolution spontanée de tout système chimique à et constantes :
- lorsque ne varie plus, est nulle[20], la fonction est minimale. Cela signifie que le système réactionnel est à l'équilibre :
À l'équilibre :
- pour une réaction chimique effectuée à et constants : ,
- la fonction énergie libre ne peut que décroître :
Condition d'évolution spontanée de tout système chimique à et constants :
- lorsque ne varie plus, est nulle, la fonction est minimale. Cela signifie que le système réactionnel est à l'équilibre :










Les relations suivantes définissent également l'affinité chimique :
| Affinité chimique : |

Équilibres hétérogènes, rupture d'équilibre
Un équilibre homogène est un équilibre dans lequel l'ensemble des réactifs et des produits se situent dans la même phase ; par exemple :
- l'équilibre du dioxyde et du trioxyde de soufre en phase gaz : ;
- l'autoprotolyse de l'eau en phase aqueuse : ;
- l'estérification en phase aqueuse : .



Un équilibre hétérogène est un équilibre dans lequel l'un au moins des réactifs ou produits se situe dans une phase différente des autres constituants du milieu réactionnel ; par exemple :
- la précipitation ou dissolution du chlorure de sodium en phases aqueuse et solide : ;
- la dissolution du dioxyde de carbone en phases gaz et aqueuse : ;
- la réduction de l'oxyde de zinc en phases solide et gaz : ;
- l'équilibre des formes allotropiques du carbone diamant et graphite : , chacun des deux solides constituant une phase à lui seul ;
- la nitration aromatique et la sulfonation aromatique faisant intervenir une phase organique liquide et une phase minérale acide liquide, les deux liquides n'étant pas miscibles.




Dans le cas des équilibres hétérogènes, si l'une des phases disparait au cours de la réaction l'état obtenu en fin de réaction n'est pas un état d'équilibre chimique : il y a rupture d'équilibre[21],[22]. D'une façon générale, tout système en équilibre chimique présentant une variance faible (1 le plus souvent) présente des ruptures d'équilibre si l'opérateur impose plus de paramètres que ne le permet la variance. Par exemple, pour un équilibre de variance 1 l'opérateur peut fixer la température mais subit la concentration. Si l'opérateur impose une concentration autre que celle dictée par l'équilibre, alors le système réactionnel, à la température fixée, est hors équilibre : il y a rupture d'équilibre. Une rupture d'équilibre peut permettre d'obtenir une réaction totale, par épuisement de l'un des réactifs[23].
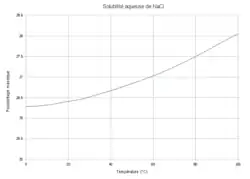
La courbe représente l'état d'équilibre, la solubilité est liée de façon univoque à la température. En dessous de la courbe de solubilité l'opérateur peut fixer indépendamment la concentration et la température : il s'agit d'une situation hors équilibre, une rupture d'équilibre.
Le chlorure de sodium (sel de table) NaCl se dissout dans l'eau selon la réaction :

Le nombre de constituants est (l'eau, NaCl, Na+ et Cl−), le nombre de réactions , le nombre de phases (solide et liquide), il existe une contrainte sur l'électroneutralité de la phase liquide (le nombre d'ions Na+ égale celui d'ions Cl−), d'où , la pression n'a pas d'influence sur l'équilibre, d'où ; la variance vaut :






L'équilibre (appelé état de saturation) est atteint lorsque les quatre espèces chimiques (en particulier le NaCl sous forme solide) sont présentes simultanément. Il ne dépend que d'un seul paramètre : si l'on fixe la température la concentration maximale en sel dissout (appelée solubilité) est subie ; réciproquement pour obtenir une solubilité donnée l'opérateur n'a pas le choix de la température. Si l'on augmente la température du sel solide se dissout et les concentrations en ions augmentent dans la solution aqueuse ; si l'on diminue la température le sel précipite et les concentrations en ions diminuent.
Toutefois, à température donnée, si la dissolution est effectuée progressivement dans de l'eau initialement pure, par ajouts successifs de petites quantités de sel, celui-ci commence par se dissoudre complètement dans l'eau et la phase solide n'est pas présente dans l'état final : la réaction est totale. Il s'agit d'une rupture d'équilibre : les conditions d'équilibre ne sont pas atteintes, il y a sous-saturation (la concentration en sel dissout est inférieure à la solubilité). Dans cet état, le nombre de constituants est (l'eau, Na+ et Cl−), le nombre de réactions (il n'y a pas équilibre), le nombre de phases (liquide seul), on a toujours la contrainte de l'électroneutralité et l'absence d'influence de la pression ; la variance vaut :




Dans cet état hors équilibre l'opérateur peut fixer la température et la concentration en sel dissout indépendamment l'une de l'autre, soit deux paramètres, un de plus que ce que l'état d'équilibre permet. L'équilibre n'est retrouvé que lorsque le sel cesse de se dissoudre : les concentrations en ions sont maximales, la solubilité est atteinte et l'opérateur perd un degré de liberté.
Le carbonate de calcium (calcaire) CaCO3 et l'oxyde de calcium (chaux) CaO sont en équilibre en présence de dioxyde de carbone CO2 selon la réaction :

La réaction est endothermique : dans un four à chaux la réaction se produit en brûlant simultanément du coke ou du charbon. La régression de la réaction est favorisée selon le principe de Berthelot (réaction exothermique) et sa progression est favorisée selon le principe de Matignon (augmentation de la quantité de constituants, création d'un gaz à partir d'un solide). Les deux réactions sont donc antagonistes, d'où l'existence de l'équilibre.
Le nombre de constituants est (CaCO3, CaO et CO2), le nombre de réactions , le nombre de phases (deux solides et un gaz), la pression et la température influencent l'équilibre, d'où , il n'y a pas d'autre contrainte, d'où ; la variance vaut :




Cet équilibre dépend de la pression du CO2. Si l'on ouvre le réacteur à l'atmosphère, alors le CO2 s'échappe du milieu réactionnel au fur et à mesure de la réaction et ne peut jamais atteindre la pression d'équilibre. La réaction va en conséquence se déplacer dans le sens de la production du CO2 jusqu'à épuisement du calcaire : la réaction est totale, il y a rupture d'équilibre, l'opérateur a imposé la température et une pression inférieure à la pression dictée par l'équilibre.
La réaction inverse est utilisée dans les absorbeurs de CO2. Si la pression de CO2 est inférieure à la pression d'équilibre, le CO2 ne réagit pas en présence de CaO et il ne se forme pas de CaCO3 : il s'agit d'une rupture d'équilibre. De même, si la pression de CO2 est supérieure à la pression d'équilibre, alors le CaO est entièrement consommé, il ne reste que du CaCO3 : la réaction est totale, il s'agit également d'une rupture d'équilibre.
Réaction à pression et température constantes
Les démonstrations qui suivent sont valables pour une réaction ayant lieu à et constantes, en se basant sur l'enthalpie libre .
Définitions
Enthalpie libre de réaction
Soient les quantités respectives des constituants de la réaction pour l'état et les enthalpies libres molaires partielles de chaque constituant à et données. L'enthalpie libre molaire partielle du constituant , qui est également son potentiel chimique, est définie par :




La différentielle de l'enthalpie libre est égale à :

Les relations liant l'évolution de l'avancement à l'évolution de chacun des constituants permettent d'écrire pour chaque constituant :
On peut donc réécrire :


L'enthalpie libre de réaction est définie par :

avec l'enthalpie libre molaire partielle ou potentiel chimique du constituant .

La différentielle de l'enthalpie libre est égale à :

d'où, à et constantes :

Cette grandeur est également notée avec l'opérateur de Lewis :

L'enthalpie libre de réaction et l'affinité chimique sont donc définies par[18],[19] :
| Enthalpie libre de réaction : |

et à pression et température constantes on a[19] :
| À et constantes : |

Enthalpie libre standard de réaction et quotient de réaction
Le potentiel chimique de chaque constituant dans le mélange réactionnel peut être exprimé en fonction du potentiel chimique de ce constituant dans un état standard à la même température que le mélange réactionnel et de l'activité du constituant selon la relation[25] :


L'état standard peut être à une autre pression, une autre composition et dans un autre état que le mélange réactionnel réel, néanmoins il est obligatoirement à la même température que celui-ci.
L'expression de l'enthalpie libre de réaction est développée selon :

Les termes standards sont regroupés dans une grandeur appelée enthalpie libre standard de réaction :
| Enthalpie libre standard de réaction : |

Les termes correspondant aux activités sont regroupés dans le quotient de réaction[25],[26] :

| Quotient de réaction : |
En conséquence l'enthalpie libre de réaction s'écrit[25] :
| Enthalpie libre de réaction : |

Constante d'équilibre
La constante d'équilibre , qui est toujours positive, est définie par la relation avec l'enthalpie standard de réaction[27] :
| Constante d'équilibre : |
La constante d'équilibre peut être déterminée expérimentalement ou calculée à partir des seules propriétés des réactifs et produits à l'état standard[27],[28],[29],[30]. Les propriétés dans l'état standard ne dépendant que de la température, la constante d'équilibre ne dépend elle-même que de la température. L'état standard est choisi à la même température que le mélange réactionnel réel (mais la pression et la composition peuvent être différentes).
L'enthalpie libre standard de réaction est liée à l'enthalpie standard de réaction et à l'entropie standard de réaction par la relation :
Lorsque et sont de même signe, peut être négative (, progression de la réaction favorisée) ou positive (, régression favorisée) : la réaction produit un équilibre d'autant plus déplacé vers la droite (vers les produits) que décroît et augmente. Nous retrouvons ici les principes de Berthelot et Matignon selon lesquels la réaction est favorisée si et : si tel est le cas, et , la réaction est totale. À contrario, la réaction est impossible si et , auquel cas et [31]. Il est considéré en pratique qu'une réaction est totale pour et impossible pour [32]. La température à laquelle et est appelée température d'inversion[33],[34] :









Elle correspond à la limite à partir de laquelle la réaction directe est favorisée ou non : pour une réaction exothermique () il vaut mieux travailler à une température inférieure à , pour une réaction endothermique () il vaut mieux travailler à une température supérieure[35].

Les relations de Kirchhoff permettent d'établir une expression rigoureuse de la constante d'équilibre :



avec :
- l'enthalpie standard de réaction à ;
- l'enthalpie molaire standard du constituant à ;
- l'entropie standard de réaction à ;
- l'entropie molaire standard du constituant à ;
- la capacité thermique isobare standard de réaction, dépendante de ;
- la capacité thermique isobare molaire standard du constituant , dépendante de ;
- la température de référence.







Sur des plages de température réduites l'enthalpie standard de réaction et l'entropie standard de réaction peuvent être considérées comme des constantes selon l'approximation d'Ellingham, ce qui revient à considérer que [36]. Aussi, en introduisant deux constantes et telles que :





on obtient[36] :

La constante d'équilibre peut être trouvée dans la littérature pour de nombreux équilibres sous la forme :

Cette expression est la plus courante, il est nécessaire de vérifier son domaine de validité en température qui est souvent assez réduit. Puisque et sont le plus souvent de même signe dans une réaction équilibrée, les constantes et sont le plus souvent de même signe.
Soit la réaction de synthèse de l'iodure d'hydrogène en phase gazeuse :

Les propriétés standards de ces espèces sont données dans le tableau suivant.
| Enthalpie standard de formation (J/mol) | Entropie molaire standard (J/(K·mol)) | Capacité thermique isobare molaire standard (J/(K·mol)) | |
|---|---|---|---|
| Hydrogène H2 | 0 | 130,684 | 28,824 |
| Iode I2 | 62 438 | 260,690 | 36,900 |
| Iodure d'hydrogène HI | 26 480 | 206,594 | 29,158 |



On calcule :
- l'enthalpie standard de réaction à 298,15 K :
- = (-1) × 0 + (-1) × 62 438 + 2 × 26 480 = −9 478 J/mol ;

- l'entropie standard de réaction à 298,15 K :
- = (-1) × 130,684 + (-1) × 260,690 + 2 × 206,594 = 21,814 J/(K·mol) ;

- la capacité thermique isobare de réaction à 298,15 K :
- = (-1) × 28,824 + (-1) × 36,900 + 2 × 29,158 = −7,408 J/(K·mol).

On a , la réaction est exothermique, et . Cette réaction devrait être totale selon Berthelot et Matignon, mais elle est équilibrée[12].


En considérant que la capacité thermique isobare de réaction, , est constante, égale à sa valeur à 298,15 K, on a par intégration des relations de Kirchhoff :

- l'enthalpie standard de réaction à :
- J/mol ;

- l'entropie standard de réaction à :
- J/mol.

On a de façon générale :


À 298,15 K on a :
- = −15 982 J/mol
- ≈ 631


À 700 K on a :
- = −12 455 J/mol
- = 15,49 J/mol
- = −23 299 J/mol
- ≈ 54,77




En considérant l'approximation d'Ellingham, on a :


À 700 K on a :
- = −9 478 J/mol
- = 21,814 J/mol
- = −24 748 J/mol
- ≈ 70,25
D'autre part, la relation de Gibbs-Helmholtz permettant d'écrire, à pression constante[27] :

on obtient la relation de van 't Hoff qui définit l'évolution de la constante d'équilibre en fonction de la température[27] :
| Relation de van 't Hoff : |

En intégrant cette relation, en supposant que l'enthalpie standard de réaction ne varie pas avec la température, on obtient :


Si l'on connait les valeurs de la constante d'équilibre et de l'enthalpie standard de réaction à la température , il est ainsi possible de déterminer la valeur de la constante d'équilibre à la température . Sous cette forme, les deux constantes et définies précédemment valent :


- ;
- .


On a donc, finalement :
En conséquence l'enthalpie libre de réaction s'écrit :
| Enthalpie libre de réaction : |

Enthalpie libre de réaction et équilibre
Sens de déplacement d'un équilibre, condition d'équilibre
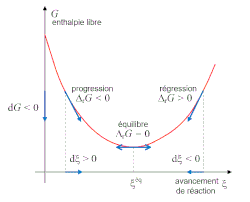
La réaction progresse lorsque . La réaction régresse lorsque . L'équilibre est atteint lorsque .
Étant donné la condition d'évolution spontanée de toute réaction et la définition de l'enthalpie libre de réaction :

on a la condition d'évolution spontanée à pression et température constantes[38] :
| Condition d'évolution spontanée à et constantes : |
Ainsi, à pression et température constantes, et ne peuvent être que de signes contraires[19] :
- et , la réaction progresse : l'équilibre se déplace de la gauche vers la droite, des réactifs sont consommés et des produits apparaissent ;
- et , la réaction régresse : l'équilibre se déplace de la droite vers la gauche, des produits sont consommés et des réactifs apparaissent.
À l'équilibre l'enthalpie libre ne varie pas, [20], d'où deux cas possibles :
- et , la réaction pourrait avoir lieu car le potentiel de réaction est non nul, mais l'avancement de réaction ne varie pas ; les causes de ce défaut de réaction peuvent être :
- une cinétique extrêmement lente (la vitesse de réaction ), l'équilibre est dit instable, c'est le cas de la transformation du diamant en graphite[alpha 6]. Un catalyseur permet d'augmenter la vitesse d'une réaction ; à contrario un inhibiteur la diminue ; dans les deux cas l'équilibre n'est pas déplacé pour autant (voir paragraphe Emploi d'un catalyseur) ;
- ou l'absence d'un facteur déclenchant (énergie, étincelle, site de nucléation, circuit de pile électrique non fermé…), l'équilibre est dit métastable[alpha 7] ;
- et , la réaction a eu lieu, le potentiel de réaction est désormais nul ; l'équilibre est dit stable ; nous ne nous intéresserons par la suite qu'à ce cas de figure.




| À l'équilibre : |
L'enthalpie libre de réaction étant liée aux enthalpie et entropie de réaction par la relation , l'état d'équilibre correspond à l'égalité , soit l'équilibre entre le facteur énergie et le facteur désordre.


Loi d'action de masse
En considérant la définition de l'enthalpie libre de réaction et la condition d'évolution spontanée à pression et température constantes :

hors équilibre nous avons :
- si alors , d'où : la réaction progresse ;
- si alors , d'où : la réaction régresse.
Lorsque la fonction est minimale, le système réactionnel est à l'équilibre ; dans ces conditions . On obtient , soit la loi d'action de masse :
| Loi d'action de masse : à l'équilibre. |

Puisque par définition et que à l'équilibre, la loi d'action de masse est vraie quelles que soient les conditions de déroulement de la réaction (pression, volume, entropie ou température constants).
Les activités chimiques qui interviennent dans le quotient de réaction dépendent de la pression , de la température et de la composition du mélange réactionnel, c'est-à-dire des quantités des réactifs et produits de réaction, mais aussi des inertes :
- .

La pression et la température , ainsi que les quantités des constituants dans le mélange réactionnel à l'instant initial (au temps ) sont des données du problème. La réaction est menée à pression et température constantes. Les quantités des constituants évoluent au cours de la réaction, hormis celles des inertes. L'équation comporte par conséquent autant d'inconnues qu'il y a de réactifs et de produits. Pour tout constituant intervenant dans la réaction, la quantité varie au cours du temps selon :
avec :
- la quantité du constituant à l'instant initial ;
- la quantité du constituant à l'instant ;
- l'avancement de réaction à l'instant ; rappelons qu'à l'instant initial, par définition, .
On peut donc remplacer dans l'expression des activités les par . Les pression, température et composition initiale étant données, le quotient de réaction devient alors à tout instant (y compris hors équilibre) une fonction de la seule variable avancement de réaction . L'équilibre est donc atteint pour l'avancement tel que :



| À l'équilibre : |

La quantité de chaque réactif et produit à l'équilibre est à postériori calculée par . L'équation n'a pas nécessairement une solution unique en . Il est entendu que sont écartées d'office les solutions mathématiques induisant des quantités de constituants négatives (rappelons toutefois que l'avancement de réaction , lui, peut être négatif). Si plusieurs solutions sont trouvées, il est nécessaire de déterminer laquelle est la bonne, ou du moins laquelle est la plus réaliste du point de vue de la thermodynamique : celle qui minimise la fonction .

On introduit dans un réacteur 0,06 mol d'hydrogénosulfure d'ammonium solide qui se décompose en ammoniac et sulfure d'hydrogène , tous deux gazeux. Le volume du réacteur est de 2,4 litres et la température est maintenue constante à 20 °C. La constante d'équilibre vaut . On écrit le tableau d'avancement, qui donne les quantités des constituants à chaque instant.




Le solide étant pur, son activité chimique vaut : . Les activités des gaz sont écrites en fonction de leur pression partielle, développée selon la loi des gaz parfaits :



avec :
- = 1 bar ;
- la constante universelle des gaz parfaits.

On a donc à l'équilibre, selon la loi d'action de masse :

On obtient = 2,2 × 10−2 mol. On complète le tableau d'avancement.

Dans cet exemple la résolution de la loi d'action de masse conduit également à la solution = −2,2 × 10−2 mol. Dans le cas présent cette solution ne peut être retenue car elle conduirait à des quantités d'ammoniac et de sulfure d'hydrogène négatives.
Condition de stabilité d'un équilibre
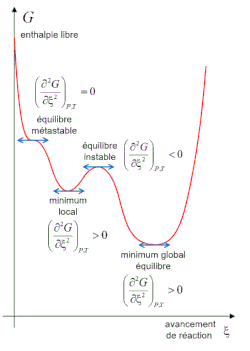

L'équilibre n'est stable que lorsque . De plus l'équilibre réel est celui pour lequel l'enthalpie libre a atteint le minimum global sur l'ensemble des valeurs possibles de l'avancement de réaction .
D'un point de vue mathématique, un minimum de est atteint si, en plus de l'annulation de sa dérivée première, sa dérivée seconde est positive strictement, soit, à l'équilibre[40],[41] :
| À l'équilibre , condition d'équilibre |

C'est à cette seconde condition seulement que l'équilibre pourra être qualifié de stable.
Il est impossible que à l'équilibre, ce qui supposerait un maximum de la fonction , et donc que a crû. Si cette solution est mathématiquement possible, elle est irréaliste du point de vue de la thermodynamique et doit donc être écartée ; mathématiquement, il s'agit d'un équilibre instable. Il est possible en revanche qu'à l'équilibre la dérivée seconde et la dérivée troisième : l'enthalpie libre a atteint un point d'inflexion et l'équilibre est un équilibre métastable, la moindre perturbation relancera la réaction jusqu'à ce qu'elle atteigne un équilibre stable. La situation est possible du point de vue de la thermodynamique. Si les dérivées seconde et troisième de l'enthalpie libre sont nulles à l'équilibre, la stabilité de l'équilibre doit être recherchée en étudiant les dérivées d'ordre supérieur. Si la première dérivée non nulle est d'ordre impair, alors l'équilibre est métastable, quel que soit son signe[alpha 8] ; si l'ordre est pair, l'équilibre est stable si cette dérivée est positive, il est instable autrement[alpha 9]. La condition de stabilité s'écrit donc de façon plus générale[40] :



| Condition de stabilité Il existe entier naturel non nul tel que , toutes les dérivées d'ordre inférieur étant nulles. |


Il est nécessaire aussi de vérifier que le minimum atteint n'est pas un minimum local, c'est-à-dire un minimum auquel la réaction revient si elle subit de petites perturbations, mais dont la réaction s'éloigne définitivement sous des perturbations plus importantes, conduisant à un autre minimum de . L'équilibre chimique à un minimum local de est un équilibre stable, néanmoins il ne s'agit pas de celui dans lequel l'enthalpie libre est minimale dans les conditions de la réaction. L'enthalpie libre peut présenter plusieurs minimums locaux. L'équilibre chimique réel est celui pour lequel l'enthalpie libre atteint le minimum global sur l'ensemble des valeurs possibles de l'avancement de réaction [alpha 10].
Les situations d'équilibre métastable ou de minimum local ne sont que rarement observées expérimentalement, mais elles peuvent être obtenues lors de simulations numériques d'équilibres. Les dérivées première et seconde de ainsi que la possibilité d'un minimum local doivent être impérativement vérifiées pour garantir le résultat. Insistons également sur le fait que les modèles d'activité chimique ou les équations d'état utilisés dans les calculs thermodynamiques restent basés sur un certain nombre d'hypothèses pouvant conduire à des résultats numériquement corrects, mais néanmoins totalement irréalistes, surtout lorsque ces modèles sont employés en dehors de leur domaine de validité.
Pour vérifier la condition de stabilité :

la constante d'équilibre ne dépendant que de la température, d'où :

Le quotient de réaction est toujours positif par définition. Pour vérifier que à l'équilibre, il faut donc vérifier que :
| Condition de stabilité de l'équilibre : |

Loi d'Arrhenius
Reprenons la convention d'écriture d'un équilibre chimique dans laquelle les coefficients stœchiométriques sont tous positifs :

On écrit, si les deux réactions sont des réactions élémentaires :
- la vitesse de la réaction directe : ;
- la vitesse de la réaction inverse : ;


avec :
- l'activité du constituant ;
- la constante de vitesse de la réaction directe exprimée selon la loi d'Arrhenius ;
- la constante de vitesse de la réaction inverse ;
- et étant les énergies d'activation respectives ;
- et étant les facteurs de fréquence respectifs.






À l'équilibre, l'égalité des vitesses des deux réactions , en notant l'activité du constituant à l'équilibre, conduit à :



Nous retrouvons ainsi le quotient de réaction à l'équilibre, noté selon la convention stœchiométrique avec les coefficients de stœchiométrie des réactifs négatifs :

Nous pouvons en conséquence à l'équilibre écrire, en considérant la loi d'action de masse établie précédemment :

d'où les relations entre loi d'Arrhenius, constante d'équilibre et enthalpie libre standard de réaction :



soit donc pour des réactions élémentaires[42],[43],[44] :
| Enthalpie standard de réaction : |
| Entropie standard de réaction : |


Récapitulatif
De façon générale, à l'équilibre et hors équilibre[45] :
- à et constantes, l'évolution spontanée de la réaction est dictée par le second principe de la thermodynamique :

- enthalpie libre de réaction :

- potentiels chimiques :
- enthalpie libre standard de réaction, potentiels chimiques à l'état standard et constante d'équilibre :

- quotient de réaction et activités :
Avec les relations :

L'enthalpie libre standard de réaction , liée à la constante d'équilibre , ne permet de définir que l'équilibre du système réactionnel. Le système réactionnel hors équilibre est étudié à l'aide de l'enthalpie libre de réaction . En effet, hors équilibre nous avons[45] :
- si , alors et : la réaction progresse ;
- si , alors et : la réaction régresse ;
et à l'équilibre[45] :
| À l'équilibre : , soit (loi d'action de masse) |
La constante d'équilibre peut être :
- calculée à partir des propriétés des réactifs et des produits dans leur état standard ;
- calculée à l'aide des paramètres de la loi d'Arrhenius des constantes dans l'expression des vitesses des réactions directe et inverse ;
- déterminée expérimentalement ;
- trouvée dans la littérature pour de nombreux équilibres.
Lors d'un calcul d'équilibre, il convient également de vérifier que la solution obtenue correspond bien à un minimum de la fonction selon la condition de stabilité :
| Condition de stabilité de l'équilibre : , soit |
Ces inégalités sont des inégalités strictes. Si la dérivée seconde de l'enthalpie libre est négative ou nulle, l'équilibre n'est pas stable et la réaction reprendra à la moindre perturbation pour atteindre un autre équilibre plus stable.
Déplacement d'un équilibre chimique
Principes généraux
Principe de modération de Le Chatelier
Soit un milieu réactionnel à l'équilibre. On modifie l'une des conditions de cet équilibre : température, pression, volume ou concentration de l'un des constituants. La réaction tend à rétablir l'équilibre, pour peu que les nouvelles conditions opératoires le permettent (pour peu que l'on ne soit pas en conditions de rupture d'équilibre). À la fin du XIXe siècle Le Chatelier (en 1884, généralisant la loi concernant la température publiée auparavant par van 't Hoff la même année) et Braun (en 1887) énoncèrent indépendamment l'un de l'autre un principe régissant ces déplacements d'équilibre[6]. Ce principe est connu sous le nom de principe de modération, ou principe de Le Chatelier ou principe de Le Chatelier-Braun, il dit que :
Le principe de Le Chatelier n'est cependant vrai que pour les modifications des paramètres intensifs qui conditionnent l'équilibre[6],[49] :
- une augmentation de température déplace l'équilibre dans le sens qui absorbe de la chaleur (endothermique) et donc qui diminue la température (loi de van 't Hoff) ;
- une augmentation de pression déplace l'équilibre dans le sens qui diminue le nombre de constituants dans le mélange et donc qui diminue la pression (loi de Le Chatelier) ;
- l'augmentation du potentiel chimique d'un constituant (par augmentation de la quantité de ce constituant - réactif, produit, inerte) déplace l'équilibre dans le sens qui diminue ce potentiel chimique.
Une augmentation du volume, qui est un paramètre extensif, induit un déplacement dans le sens de l'augmentation du volume du milieu réactionnel, et non de sa diminution. Toutefois, le principe de Le Chatelier est respecté si l'on considère la pression de ce système, qui est diminuée par l'augmentation du volume et augmentée par la réaction[6].
Pour un équilibre dans lequel on modifie la composition par ajout ou extraction d'un constituant (réactif, produit, inerte), le principe de Le Chatelier peut ne pas être respecté si l'on considère la quantité de ce constituant, qui est un paramètre extensif. Il existe en effet des contre-exemples dans lesquels, sous certaines conditions, la réaction régresse lors d'un ajout de réactif, au lieu de progresser : la réaction augmente la quantité du réactif ajouté au lieu de la diminuer[4],[5]. Toutefois, le principe de Le Chatelier est respecté si l'on considère le potentiel chimique de ce constituant, qui est augmenté par l'ajout du constituant et diminué par la réaction[6].
Principe général d'étude du déplacement d'un équilibre chimique
Soit un milieu réactionnel à l'équilibre. Son affinité chimique initiale est donc nulle :

On modifie l'une des conditions opératoires (par exemple la pression, la température ou la quantité de l'un des constituants).

Dans un premier temps, on considère que le milieu réactionnel est modifié en l'absence de réaction, il se trouve donc hors équilibre chimique après modification de . Cette condition devient , les autres conditions opératoires restant constantes. L'affinité chimique varie de :



et devient :

On considère ensuite que la réaction débute seulement après cette modification. Le deuxième principe de la thermodynamique induit que la réaction se déplace dans le sens tel que[50],[51] :

On obtient la relation qui détermine le sens de déplacement de la réaction[52] :
| Condition de déplacement de l'équilibre : |

À constantes, on modifie la valeur de , soit . Si , l'affinité chimique est modifiée et n'est plus nulle ; le système se trouve hors équilibre et la réaction se déplace pour retrouver l'équilibre, soit . L'étude du déplacement de la réaction consiste donc :


- à établir, dans les conditions de l'équilibre initial, l'expression de et son signe ;
- à modifier la condition opératoire , ce qui revient à imposer le signe de ;
- à déduire le signe de de la condition d'évolution spontanée, et donc le sens de déplacement de la réaction.


Si et sont de même signe, alors ne peut être que positif et la réaction ne peut que progresser. Inversement, s'ils sont de signes opposés alors la réaction ne peut que régresser.
Si on étudie les différentielles d'ordre supérieur. En supposant que l'affinité est fois dérivable, le développement en série de Taylor donne :


avec la différentielle d'ordre de à constantes. On étudie le premier ordre auquel à l'équilibre. On a alors :




d'où la condition de déplacement de l'équilibre :



Il se peut que n'existe pas, autrement dit que quel que soit on ait : dans ce cas l'équilibre est neutre, totalement insensible à la modification de . Par exemple, l'ajout ou l'extraction du composant de la phase hétérogène d'un équilibre n'a aucune influence sur l'équilibre. De même, un équilibre est insensible à l'ajout ou l'extraction d'un inerte à volume et température constants. Si est impair, alors le signe de dépend du signe de : l'augmentation et la diminution de ont des conséquences inverses sur la réaction. Par exemple, l'augmentation et la diminution de la température, ou de la pression, déplacent l'équilibre dans des sens opposés. Si est pair, alors le signe de ne dépend pas du signe de : la réaction se déplace dans le même sens quelle que soit la modification de . Par exemple, dans certaines conditions de composition et de stœchiométrie, à pression et température constantes, la réaction régresse que l'on ajoute ou extraie l'un des réactifs.
Principes généraux de déplacement de l'équilibre
Selon les conditions opératoires, on considère le potentiel thermodynamique , fonction des variables , tel que :


Soit la variable conjuguée de :


Note - La « variable conjuguée » s'entend ici au sens de dérivée partielle du potentiel thermodynamique par rapport à la variable , ce qui implique de tenir compte de son signe. Ainsi, au sens de l'enthalpie libre , la variable conjuguée de la pression est le volume, . Mais au sens de l'énergie libre , la variable conjuguée du volume est l'opposé de la pression, .


Le théorème de Schwarz donne :

On note la variation de due à la réaction dans les conditions constantes :


La condition de déplacement de l'équilibre devient finalement :


On en déduit que :
| Une augmentation de déplace la réaction dans le sens impliquant la diminution de sa variable conjuguée . | Une diminution de déplace la réaction dans le sens impliquant l'augmentation de sa variable conjuguée . |
On note la variation de en l'absence de réaction, dans les conditions constantes :

La variable conjuguée varie en fonction de selon[alpha 11] :


En l'absence de réaction, l'augmentation d'une variable intensive fait diminuer sa variable conjuguée extensive, et l'augmentation d'une variable extensive fait augmenter sa variable conjuguée intensive.
Le milieu réactionnel évolue à la fois par la modification externe de et par le déplacement interne de l'équilibre, ces deux évolutions pouvant aller dans le même sens ou s'opposer. On peut alors comparer les évolutions de en l'absence et en présence de la réaction :


On en déduit que :
- si est intensive, sa variable conjuguée est extensive :
- une augmentation de , soit , induit que et ;
- une diminution de , soit , induit que et ;
- la variation de est plus importante avec la réaction que sans la réaction : ;
- si est extensive, sa variable conjuguée est intensive :
- une augmentation de , soit , induit que et ;
- une diminution de , soit , induit que et ;
- la variation de est moins importante avec la réaction que sans la réaction : .








En conséquence, le milieu réactionnel évolue selon les principes généraux suivants[6],[53],[54] :
| La modification d'une variable intensive induit une variation de sa variable conjuguée extensive plus grande avec la réaction que sans la réaction. Autrement dit, la modification de déplace l'équilibre dans le sens qui amplifie la modification de . |
| La modification d'une variable extensive induit une variation de sa variable conjuguée intensive plus petite avec la réaction que sans la réaction. Autrement dit, la modification de déplace l'équilibre dans le sens qui s'oppose à la modification de . |
Le tableau suivant résume les modifications de conditions opératoires les plus courantes d'un équilibre chimique[55].
| Modification | Conditions opératoires constantes | Évolution | |||||
|---|---|---|---|---|---|---|---|
| Condition opératoire | Opération | Potentiel thermodynamique | Variable conjuguée | Par la réaction | Sans la réaction | Avec la réaction | |
| augmentation | diminue (augmente) | diminue (augmente) | diminue plus (augmente plus) | ||||
| diminution | augmente (diminue) | augmente (diminue) | augmente plus (diminue plus) | ||||
| augmentation | diminue (augmente) | diminue (augmente) | diminue plus (augmente plus) | ||||
| diminution | augmente (diminue) | augmente (diminue) | augmente plus (diminue plus) | ||||
| augmentation | diminue | diminue | diminue plus | ||||
| diminution | augmente | augmente | augmente plus | ||||
| augmentation | diminue (augmente) | augmente (diminue) | augmente moins (diminue moins) | ||||
| diminution | augmente (diminue) | diminue (augmente) | diminue moins (augmente moins) | ||||
| augmentation | et | diminue | augmente | augmente moins | |||
| diminution | augmente | diminue | diminue moins | ||||
| augmentation | et | diminue | augmente | augmente moins | |||
| diminution | augmente | diminue | diminue moins | ||||









Avec :
- l'énergie libre ;
- l'enthalpie libre ;
- la quantité du constituant (réactif, produit, inerte) - variable extensive ;
- la pression - variable intensive ;
- l'entropie du milieu réactionnel - variable extensive ;
- la température - variable intensive ;
- le volume du milieu réactionnel - variable extensive ;
- le potentiel chimique du constituant (réactif, produit ou inerte) - variable intensive.

Modification de la température
Condition d'évolution spontanée par modification de la température
Supposons un équilibre chimique à une condition (volume ou pression) et à température . La température est modifiée à , la condition restant constante. On a la variation d'affinité chimique[6],[49] :


On obtient la relation qui détermine le sens de déplacement de la réaction par modification de la température :
| Condition d'évolution spontanée par modification de la température : |

avec l'entropie du milieu réactionnel.
Soit le potentiel thermodynamique . Si le paramètre est le volume il s'agit de l'énergie libre , si est la pression il s'agit de l'enthalpie libre . On a par définition de l'affinité chimique :




Le théorème de Schwarz permet d'écrire :

On a la relation entre énergie libre, enthalpie libre et entropie :

d'où :
Cas de la modification de la température à pression constante
À pression constante on a la variation d'affinité chimique[56],[57] :

On obtient la relation qui détermine le sens de déplacement de la réaction par modification de la température à pression constante :

avec :
- l'enthalpie de réaction ;
- l'enthalpie molaire partielle du constituant .


La variation d'affinité engendrée par la variation de température à pression constante vaut :

Puisque l'on travaille avec les paramètres pression et température, le potentiel thermodynamique le plus approprié pour l'étude est l'enthalpie libre . Par définition , avec l'enthalpie. À l'état initial, avant modification de la température, l'équilibre chimique donne :




On a par conséquent à l'équilibre :

Il vient cinq cas de figure[56],[57],[58] :
- , la réaction est athermique[59], elle ne dégage ni n'absorbe aucune chaleur, d'où ; l'affinité chimique n'est pas modifiée par la modification de la température, elle reste donc nulle : l'équilibre est neutre, insensible à la température, et ne se déplace pas (en toute rigueur, il faut étudier les dérivées d'ordre supérieur de par rapport à la température) ;
- , la réaction est exothermique[60], la réaction dégage de la chaleur en progressant :
- si l'on augmente la température : , d'où , la réaction régresse ;
- si l'on diminue la température : , d'où , la réaction progresse ;
- , la réaction est endothermique[61], la réaction absorbe de la chaleur en progressant :
- si l'on augmente la température : , d'où , la réaction progresse ;
- si l'on diminue la température : , d'où , la réaction régresse.






La variation de l'énergie interne du milieu réactionnel est égale à la somme du travail et de la chaleur reçus par ce milieu, soit, en termes élémentaires :




Si le seul travail est celui des forces de pression, alors . À pression constante on a :

![{\displaystyle \mathrm {d} U+P\,\mathrm {d} V=\mathrm {d} \left[U+PV\right]=\mathrm {d} H=\delta Q_{P}}](https://img.franco.wiki/i/414ed0d0e175de0a8473cca7add5ed8cfba0932e.svg)
avec l'enthalpie. À pression constante la variation de l'enthalpie est égale à la chaleur échangée par le système avec l'extérieur. On peut écrire[58] :


Par conséquent, à pression et température constantes[62] :

Par définition, une réaction qui progresse, soit , est :
- athermique si elle ne dégage aucune chaleur[59], soit ;
- exothermique si elle dégage de la chaleur[60], soit ;
- endothermique si elle absorbe de la chaleur[61], soit .



On en déduit que, à pression et température constantes :
- si la réaction est exothermique ;
- si la réaction est endothermique.
Cas de la modification de la température à volume constant
À volume constant on a la variation d'affinité chimique[6],[49] :

On obtient la relation qui détermine le sens de déplacement de la réaction par modification de la température à volume constant :
![{\displaystyle \left[\Delta _{\text{r}}H-l\,\Delta _{\text{r}}V\right]\,{\mathrm {d} T \over T}\,\mathrm {d} \xi \geq 0}](https://img.franco.wiki/i/fbde51ce891b938da1e5a086e80b76ce98f108cc.svg)
avec :
- le coefficient de dilatation isotherme du milieu réactionnel, donné par la première relation de Clapeyron ;
- l'enthalpie de réaction ;
- l'enthalpie molaire partielle du constituant ;
- le volume de réaction ;
- le volume molaire partiel du constituant .



La variation d'affinité engendrée par la variation de température à volume constant vaut :

Puisque l'on travaille avec les paramètres volume et température, le potentiel thermodynamique le plus approprié pour l'étude est l'énergie libre . Par définition , avec l'énergie interne. À l'état initial, avant modification de la température, l'équilibre chimique donne :



On a la relation :

dans laquelle on introduit la relation liant l'énergie interne et le coefficient de dilatation isotherme :


et l'enthalpie :
![{\displaystyle \Delta _{\text{r}}U=\left({\partial U \over \partial \xi }\right)_{P,T}=\left({\partial \left[H-PV\right] \over \partial \xi }\right)_{P,T}=\left({\partial H \over \partial \xi }\right)_{P,T}-P\,\left({\partial V \over \partial \xi }\right)_{P,T}=\Delta _{\text{r}}H-P\,\Delta _{\text{r}}V}](https://img.franco.wiki/i/1f37ed9a16b0954fdf750f33e19123a8f82a1418.svg)
On obtient par conséquent :

d'où, à l'équilibre :

Il vient cinq cas de figure[6],[49] :
- , la réaction est athermique[59], elle ne dégage ni n'absorbe aucune chaleur, d'où ; l'affinité chimique n'est pas modifiée par la modification de la température, elle reste donc nulle : l'équilibre est neutre, insensible à la température, et ne se déplace pas (en toute rigueur, il faut étudier les dérivées d'ordre supérieur de par rapport à la température) ;
- , la réaction est exothermique[60], la réaction dégage de la chaleur en progressant :
- si l'on augmente la température : , d'où , la réaction régresse ;
- si l'on diminue la température : , d'où , la réaction progresse ;
- , la réaction est endothermique[61], la réaction absorbe de la chaleur en progressant :
- si l'on augmente la température : , d'où , la réaction progresse ;
- si l'on diminue la température : , d'où , la réaction régresse.




La variation de l'énergie interne du milieu réactionnel est égale à la somme du travail et de la chaleur reçus par ce milieu, soit, en termes élémentaires :
Si le seul travail est celui des forces de pression, alors . Par conséquent, à volume constant, soit , on a et pour l'énergie interne :



La variation de l'énergie interne à volume constant est égale à la chaleur échangée par le système avec l'extérieur. On peut écrire :

Par conséquent, à volume et température constants[62] :

Par définition, une réaction qui progresse, soit , est :
- athermique si elle ne dégage aucune chaleur[59], soit ;
- exothermique si elle dégage de la chaleur[60], soit ;
- endothermique si elle absorbe de la chaleur[61], soit .
On en déduit que, à volume et température constants :
- si la réaction est exothermique ;
- si la réaction est endothermique.
Cas général pour la modification de la température, loi de van 't Hoff
Les deux cas sont résumés par la loi de van 't Hoff, qui la découvrit expérimentalement :
| Loi de van 't Hoff : une augmentation de température à pression ou volume constant déplace l'équilibre dans le sens endothermique[41],[63]. | Inversement, une diminution de température à pression ou volume constant déplace l'équilibre dans le sens exothermique. |
Afin de comprendre en quoi le principe de Le Chatelier est respecté, imaginons un réacteur à la température auquel on communique une certaine chaleur. En l'absence de réaction on obtiendrait une température . Cependant la réaction se déplace et, puisque la température a augmenté, elle absorbe de la chaleur (déplacement dans le sens endothermique) ; lorsque l'équilibre se stabilise on obtient une température . Pour une même chaleur fournie au système, en l'absence de réaction toute la chaleur sert à chauffer le milieu réactionnel, tandis que la réaction absorbe une partie de cette chaleur, qui n'est donc pas transmise en totalité au milieu. La température en l'absence de réaction est donc supérieure à la température avec la réaction : . On obtient finalement une température plus faible que prévu : la réaction s'est opposée à l'augmentation de la température.



L'estérification consiste en la production d'un ester et d'eau à partir d'un acide carboxylique et d'un alcool selon la réaction :




Cette réaction est quasiment athermique : cet équilibre est insensible aux changements de température.

Ampoules de dioxyde d'azote portées respectivement, de gauche à droite, à : −196 °C, 0 °C, 23 °C, 35 °C et 50 °C. À basse température le dimère N2O4, incolore, est prépondérant. À haute température c'est au contraire le monomère NO2, brun, qui est prépondérant. Plus la température augmente, plus l'équilibre se déplace vers la production de monomère et plus le contenu de l'ampoule devient foncé.
Le monomère dioxyde d'azote NO2 et son dimère le peroxyde d'azote N2O4 sont en équilibre permanent selon la réaction :

La chaleur de réaction est de (298,15 K) = −57,23 kJ/mol, la réaction est donc exothermique lors d'un déplacement de la gauche vers la droite. En conséquence, plus la température augmente, plus l'équilibre se déplace de la droite vers la gauche (sens endothermique). À basse température, le dimère N2O4 est l'espèce prépondérante, à haute température c'est le monomère NO2.
Nous vérifions d'autre part que la progression de la réaction est favorisée selon le principe de Berthelot (réaction exothermique), et que la régression de la réaction est favorisée selon le principe de Matignon (augmentation de la quantité de constituants). Les deux réactions sont donc antagonistes d'où l'existence de l'équilibre.
Modification de la pression ou du volume
Modification de la pression
Condition d'évolution spontanée par modification de la pression
Supposons un équilibre chimique à pression et température . La pression est modifiée à , la température restant constante. On a la variation d'affinité chimique[41],[57] :


On obtient la relation qui détermine le sens de déplacement de la réaction par modification de la pression :
| Condition d'évolution spontanée par modification de la pression : |

avec :
- le volume de réaction ;
- le volume molaire partiel du constituant .
Puisque l'on travaille avec les paramètres pression et température, le potentiel thermodynamique le plus approprié pour l'étude est l'enthalpie libre . On a par définition de l'affinité chimique :

Le théorème de Schwarz permet d'écrire :

On a la relation entre enthalpie libre et volume :

d'où :

Cas général pour la modification de la pression
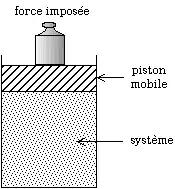
Modification de la pression par les poids posés sur le piston. Si l'on augmente la pression, le volume diminue plus avec la réaction que sans la réaction. Pour obtenir un certain volume, il faut donc appliquer une pression moins forte avec la réaction que sans la réaction.
Il vient cinq cas de figure[41] :
- , la réaction ne modifie pas le volume du milieu réactionnel, d'où ; l'affinité chimique n'est pas modifiée par la modification de la pression, elle reste donc nulle : l'équilibre est neutre, insensible à la pression, et ne se déplace pas (en toute rigueur, il faut étudier les dérivées d'ordre supérieur de par rapport à la pression) ;
- , le volume du milieu réactionnel diminue lorsque la réaction progresse :
- si l'on augmente la pression : , d'où , la réaction progresse ;
- si l'on diminue la pression : , d'où , la réaction régresse ;
- , le volume du milieu réactionnel augmente lorsque la réaction progresse :
- si l'on augmente la pression : , d'où , la réaction régresse ;
- si l'on diminue la pression : , d'où , la réaction progresse.






Pour résumer :
| Une augmentation de pression à température constante déplace l'équilibre dans le sens dans lequel le volume du milieu réactionnel diminue[41]. | Inversement, une diminution de pression à température constante déplace l'équilibre dans le sens dans lequel le volume du milieu réactionnel augmente. |
Afin de comprendre en quoi le principe de Le Chatelier est respecté, imaginons un mélange réactionnel gazeux dans un piston. Soit la pression initiale. Pour modifier la pression, on pose des poids sur le piston. Soit le volume que l'on atteindrait en l'absence de réaction chimique, avec la pression . Cependant la réaction se déplace et, puisque la pression a augmenté, tend à diminuer le volume. Soit le volume que l'on atteint avec la réaction chimique. On a par conséquent à . On obtient finalement un volume plus faible que prévu : la réaction a amplifié la diminution du volume.





Si, plutôt que de modifier la pression, on modifie le volume du piston, on impose , auquel on souhaiterait obtenir la pression en l'absence de réaction. On obtient, avec la réaction, une pression à . On obtient finalement une pression plus faible que prévu : la réaction s'est opposée à l'augmentation de la pression.


Le graphite et le diamant sont deux formes allotropiques du carbone en équilibre selon :

Le volume molaire du diamant est de 3,416 6 cm3/mol, le volume molaire du graphite est de 5,298 2 cm3/mol. Pour les solides, il peut être considéré que le volume molaire partiel est égal au volume molaire du corps pur. Le volume de réaction est donc de : = –5,2982 + 3,4166 = −1,881 6 cm3/mol. La synthèse du diamant est effectuée à haute pression : 58 000 atm. Revenu à basse pression, le diamant tend à redevenir graphite, mais la réaction est extrêmement lente, aussi parle-t-on d'équilibre instable.

Cas des gaz parfaits pour la modification de la pression, loi de Le Chatelier
Pour les gaz parfaits on a la variation d'affinité chimique[67] :

On obtient la relation qui détermine le sens de déplacement de la réaction par modification de la pression :

avec la somme des coefficients stœchiométriques de la réaction, notés selon la convention stœchiométrique (coefficients des réactifs négatifs).

Pour les gaz parfaits, le volume total vaut, selon la loi des gaz parfaits :

le volume molaire partiel de chacun des constituants vaut :

d'où :

Il vient cinq cas de figure[67] :
- , la réaction ne modifie pas la quantité totale de constituants (réactifs et produits) ; on a, pour tout entier naturel non nul, ; l'affinité chimique n'est pas modifiée par la modification de la pression, elle reste donc nulle : l'équilibre est neutre, insensible à la pression, et ne se déplace pas ;
- , la quantité totale de constituants (réactifs et produits) diminue lorsque la réaction progresse :
- si l'on augmente la pression : , d'où , la réaction progresse ;
- si l'on diminue la pression : , d'où , la réaction régresse ;
- , la quantité totale de constituants (réactifs et produits) augmente lorsque la réaction progresse :
- si l'on augmente la pression : , d'où , la réaction régresse ;
- si l'on diminue la pression : , d'où , la réaction progresse.




Ces relations sont résumées par la loi de Le Chatelier, qui la découvrit expérimentalement :
| Loi de Le Chatelier : une augmentation de pression à température constante déplace l'équilibre dans le sens qui diminue la quantité totale de constituants (réactifs et produits) dans le mélange[41]. | Inversement, une diminution de pression à température constante déplace l'équilibre dans le sens qui augmente la quantité totale de constituants (réactifs et produits) dans le mélange. |
En augmentant la pression la réaction diminue la quantité de constituants, ce qui tend à diminuer la pression : la réaction s'oppose donc à la modification de pression.
La réaction de synthèse de l'iodure d'hydrogène à partir d'hydrogène et d'iode s'écrit :

La somme des coefficients stœchiométriques est de = –1 –1 +2 = 0. Cette réaction est insensible aux changements de pression.

Dans le procédé Haber-Bosch, la synthèse de l'ammoniac est effectuée selon la réaction :

La somme des coefficients stœchiométriques est de = –1 –3 +2 = –2. La synthèse de l'ammoniac est effectuée à haute pression, entre 150 et 250 bar.
Modification du volume
Condition d'évolution spontanée par modification du volume
Supposons un équilibre chimique à volume et température . Le volume est modifié à , la température restant constante. On a la variation d'affinité chimique :


On obtient la relation qui détermine le sens de déplacement de la réaction par modification du volume :
| Condition d'évolution spontanée par modification du volume : |

avec :
- le volume de réaction ;
- le volume molaire partiel du constituant ;
- le coefficient de compressibilité isotherme.

Puisque l'on travaille avec les paramètres volume et température, le potentiel thermodynamique le plus approprié pour l'étude est l'énergie libre . On a par définition de l'affinité chimique :

Le théorème de Schwarz permet d'écrire :

On a la relation entre énergie libre et pression :

d'où :

La relation entre dérivées partielles :

permet d'écrire finalement :

La dérivée à avancement de réaction constant, c'est-à-dire en l'absence de réaction, revient à la dérivée à composition constante :

Cas général pour la modification du volume
Modification du volume par déplacement du piston. Si l'on diminue le volume, la pression augmente moins avec la réaction que sans la réaction. Pour obtenir une certaine pression, il faut donc diminuer plus fortement le volume avec la réaction que sans la réaction.
Le deuxième principe de la thermodynamique implique qu'un système thermodynamique ne peut être stable que si son volume diminue lorsque la pression augmente, soit pour le coefficient de compressibilité isotherme (voir l'article Compressibilité) :

On a par ailleurs, à température et composition constantes :

On retrouve la condition d'évolution spontanée par modification de la pression :
En conséquence, si l'on diminue le volume du milieu réactionnel, soit , l'équilibre se déplace dans le sens déterminé par une augmentation de pression, soit , c'est-à-dire le sens dans lequel le volume du milieu réactionnel diminue (voir paragraphe Cas général pour la modification de la pression). Ainsi, contrairement à ce que prédit le principe de Le Chatelier, la réaction ne se déplace pas dans le sens qui s'oppose à la diminution du volume, au contraire, elle accompagne ce changement de volume. En revanche, elle s'oppose à l'augmentation de la pression[6],[68].


Le déplacement de l'équilibre est conforme au principe de Le Chatelier au regard de la pression.
| Une augmentation de volume à température constante déplace l'équilibre dans le sens qui augmente la pression. | Inversement, une diminution de volume à température constante déplace l'équilibre dans le sens qui diminue la pression. |
Au regard du volume, les conclusions sont donc l'inverse de celles du principe de Le Chatelier : la réaction accompagne la modification du volume.
Afin de comprendre en quoi le principe de Le Chatelier est respecté, imaginons un mélange réactionnel gazeux dans un piston. Soit le volume initial. Pour modifier le volume, on déplace le piston. Soit la pression que l'on atteindrait en l'absence de réaction chimique, avec le volume . Cependant la réaction se déplace et, puisque le volume a diminué, tend à diminuer la pression. Soit la pression que l'on atteint avec la réaction chimique. On a par conséquent à . On obtient finalement une pression plus faible que prévu : la réaction s'est opposé à l'augmentation de pression.


Si, plutôt que de modifier le volume, on modifie la pression en posant des poids sur le piston, on impose , à laquelle on souhaiterait obtenir le volume en l'absence de réaction. On obtient, avec la réaction, un volume à . On obtient finalement un volume plus faible que prévu : la réaction a amplifié la diminution du volume.
Cas des gaz parfaits pour la modification du volume
Pour les gaz parfaits on a la variation d'affinité chimique :

On obtient la relation qui détermine le sens de déplacement de la réaction par modification du volume :

avec la somme des coefficients stœchiométriques de la réaction, notés selon la convention stœchiométrique (coefficients des réactifs négatifs).
Le volume total d'un mélange de gaz parfaits vaut, selon la loi des gaz parfaits :
Le coefficient de compressibilité isotherme vaut :

Le volume molaire partiel de chacun des constituants vaut :
d'où :
Il vient cinq cas de figure[68] :
- , la réaction ne modifie pas la quantité totale de constituants (réactifs et produits) ; on a, pour tout entier naturel non nul, ; l'affinité chimique n'est pas modifiée par la modification du volume, elle reste donc nulle : l'équilibre est neutre, insensible au volume, et ne se déplace pas ;
- , la quantité totale de constituants (réactifs et produits) diminue lorsque la réaction progresse :
- si l'on augmente le volume : , d'où , la réaction régresse ;
- si l'on diminue le volume : , d'où , la réaction progresse ;
- , la quantité totale de constituants (réactifs et produits) augmente lorsque la réaction progresse :
- si l'on augmente le volume : , d'où , la réaction progresse ;
- si l'on diminue le volume : , d'où , la réaction régresse.


Pour résumer :
| Une augmentation du volume à température constante déplace l'équilibre dans le sens qui augmente la quantité totale de constituants (réactifs et produits) dans le mélange[68]. | Inversement, une diminution du volume à température constante déplace l'équilibre dans le sens qui diminue la quantité totale de constituants (réactifs et produits) dans le mélange. |
Modification de la composition
Condition d'évolution spontanée par modification de la composition
Supposons un équilibre chimique à une condition (volume ou pression) et à température . La quantité du constituant (réactif, produit, inerte) est modifiée à , la condition et la température restant constantes. On a par définition l'affinité chimique :

et par conséquent sa variation[69] :

On obtient la relation qui détermine le sens de déplacement de la réaction par modification de la composition :
| Condition d'évolution spontanée par modification de la composition : |

avec :
- la quantité du constituant (réactif, produit, inerte) ;
- la pression ou le volume ;
- le potentiel chimique du constituant .
Une étude plus fine est complexe. Elle nécessite de connaitre l'évolution des potentiels chimiques en fonction de la composition du milieu réactionnel, ce qui requiert l'utilisation d'un modèle d'activité chimique. La relation (voir le paragraphe Constante d'équilibre) :

avec :
- l'enthalpie libre de réaction ;
- le quotient de réaction ; étant l'activité chimique du constituant ;
- la constante de réaction, ne dépendant que de la température ;
donne :

Cette étude peut être simplifiée dans le cas des solutions idéales, incluant les mélanges de gaz parfaits.
Cas général pour la modification de composition, respect du principe de Le Chatelier
On note de façon générique un potentiel thermodynamique . Si la condition est le volume , il s'agit de l'énergie libre . Si la condition est la pression , il s'agit de l'enthalpie libre . On a les relations avec l'affinité chimique et le potentiel chimique :

En application du théorème de Schwarz :

La condition d'évolution spontanée par modification de la composition peut donc également s'écrire :

On note la variation du potentiel chimique du constituant due à la réaction dans les conditions et constantes :

On obtient finalement[6] :

Il vient trois cas de figure :
- , la réaction ne modifie pas le potentiel chimique du constituant : l'équilibre est insensible à la quantité de et ne se déplace pas que l'on ajoute ou extraie ce constituant ;
- si l'on ajoute le constituant : , d'où , la réaction diminue son potentiel chimique ;
- si l'on extrait le constituant : , d'où , la réaction augmente son potentiel chimique.





Pour résumer :
| Une augmentation de la quantité d'un constituant (réactif, produit, inerte) déplace l'équilibre dans le sens qui diminue le potentiel chimique de ce constituant. | Inversement, une diminution de la quantité d'un constituant (réactif, produit, inerte) déplace l'équilibre dans le sens qui augmente le potentiel chimique de ce constituant. |
On n'en tire aucune conclusion quant au sens de déplacement de la réaction (le signe de ), qui peut donc aussi bien progresser que régresser pour atteindre ce résultat. Le principe de modération voudrait que d'une façon générale l'ajout d'un réactif (ou l'extraction d'un produit) déplace l'équilibre dans le sens de la consommation du réactif (respectivement de l'apparition du produit), donc que la réaction progresse en diminuant la quantité du réactif ajouté (en augmentant la quantité du produit extrait). À contrario, l'extraction d'un réactif ou l'ajout d'un produit devrait avoir pour conséquence une régression de la réaction. Il n'en va pas toujours ainsi dans les faits. Il existe des cas où il ne se passe rien et des cas où l'effet inverse se produit, par exemple où l'ajout d'un réactif induit la régression de la réaction, et non sa progression : la réaction augmente la quantité du réactif ajouté. Ceci se démontre dans le cas des équilibres hétérogènes et dans le cas des équilibres en solution idéale. Ainsi, au regard de la quantité du constituant, le principe de Le Chatelier n'est pas toujours observé lors de l'ajout ou de l'extraction de l'un des constituants du mélange réactionnel[4],[5].
Le principe de Le Chatelier est toutefois respecté si l'on considère non pas la quantité du constituant que l'on ajoute ou extrait, mais son potentiel chimique. Le deuxième principe de la thermodynamique induit en effet que le mélange de constituants, en l'absence de réaction, ne peut être stable que si[6] :

De façon générale, quel que soit le signe de la modification , on a donc :


On note la variation du potentiel chimique due à la modification de la quantité du constituant (réactif, produit, inerte) en maintenant , et les quantités des autres constituants constantes (en l'absence de réaction)[6] :


On obtient finalement :

Par conséquent, dans les conditions et constantes et en l'absence de réaction :
- si l'on ajoute le constituant : , d'où , son potentiel chimique augmente ;
- si l'on extrait le constituant : , d'où , son potentiel chimique diminue.


La modification de la quantité provoque une modification totale du potentiel chimique égale à :

Ainsi, si l'on augmente la quantité du constituant , soit , on a et . L'ajout du constituant augmente son potentiel chimique, tandis que la réaction le diminue. Inversement, l'extraction du constituant diminue son potentiel chimique, tandis que la réaction l'augmente. Le principe de Le Chatelier est donc respecté au regard du potentiel chimique du constituant dont on modifie la quantité, et non au regard de cette quantité : la réaction s'oppose à la modification des potentiels chimiques des constituants du milieu réactionnel, y compris les inertes[6].

Cas des équilibres hétérogènes
On considère un équilibre hétérogène dont la réaction chimique a lieu dans une phase et implique une espèce pure dans une phase (par exemple, dans une réaction de précipitation, un solide produit par une réaction en phase liquide) :



L'affinité chimique vaut :

On modifie la quantité d'un constituant quelconque à pression et température constantes, d'où :

Si le constituant est l'espèce , celle-ci étant pure dans la phase , son potentiel chimique ne dépend pas de la composition ; on a par conséquent, à pression et température constantes :


La quantité de l'espèce en phase n'intervient pas dans les expressions des potentiels chimiques des autres espèces dans le milieu réactionnel , ces potentiels ne dépendant que de la pression, de la température et de la composition de la phase . On a par conséquent, à pression et température constantes :


Aussi, à pression et température constantes, si l'on fait varier la quantité de l'espèce dans la phase , on a[69] :

On a également, pour tout entier naturel non nul :

En conclusion, l'affinité chimique n'est pas modifiée par la modification de la quantité de , elle reste donc nulle ; l'équilibre est neutre, insensible à la quantité de , et ne se déplace pas :
| À pression et température constantes un équilibre hétérogène est insensible à l'ajout ou l'extraction de la phase hétérogène au milieu réactionnel[69],[70]. |

Un marais salant est le siège d'un équilibre hétérogène entre un milieu réactionnel liquide, la saumure, et un milieu solide, le sel cristallisé. L'évaporation de l'eau augmente la concentration du sel dissout dans la saumure, ce qui provoque la précipitation du sel. En revanche, l'extraction du sel solide n'a aucune influence sur la concentration du sel dissout dans la saumure.
En phase aqueuse le chlorure de sodium, ou sel de table, se dissout selon la réaction :
Le quotient de réaction s'écrit :

Le sel solide est considéré comme un corps pur. Son état standard est pris comme étant le sel pur solide à la même température que le milieu réactionnel. Son activité vaut 1 : . Pour les deux ions, l'état standard considéré est l'ion à une concentration de 1 mol/l en solution aqueuse à la même température que le milieu réactionnel. Les activités des deux ions sont assimilées à leur concentration respective : et . On retrouve le produit de solubilité de la réaction de dissolution à saturation :


![{\displaystyle a_{\rm {Na^{+}}}^{\rm {aq}}=\left[{\rm {Na^{+}}}\right]/c^{\circ }=\left[{\rm {Na^{+}}}\right]}](https://img.franco.wiki/i/8c320148bd36aacbde32b41429ba22a4b90a899e.svg)
![{\displaystyle a_{\rm {Cl^{-}}}^{\rm {aq}}=\left[{\rm {Cl^{-}}}\right]/c^{\circ }=\left[{\rm {Cl^{-}}}\right]}](https://img.franco.wiki/i/afc07dc71dcd2fd55ac82dbf706ad871ddc456d9.svg)
![{\displaystyle K=\left[{\rm {Na^{+}}}\right]\cdot \left[{\rm {Cl^{-}}}\right]}](https://img.franco.wiki/i/6b9dadd7b18bd65824b4ee1ff70610ebea2c6f04.svg)
Lorsque la saturation est atteinte, l'ajout ou l'extraction de sel en phase solide n'a aucune influence sur la concentration des ions en phase aqueuse : l'équilibre n'est pas modifié.
En revanche, l'ajout de l'un des deux ions (par exemple de Na+ par l'intermédiaire d'un autre sel comme le sulfate de sodium Na2SO4 ou de Cl− par l'intermédiaire de chlorure d'hydrogène HCl) en phase aqueuse provoque une modification de l'équilibre, favorisant la précipitation du NaCl. Au contraire l'extraction de l'un des deux ions favorise la dissolution du NaCl. C'est l'effet d'ion commun. De même, si l'on ajoute de l'eau on dilue les ions et l'on favorise la dissolution du sel ; si l'on évapore l'eau on concentre les ions et le sel précipite (c'est le principe du marais salant).
En conclusion, la variation de la quantité de sel solide, dans la phase hétérogène donc, n'a aucune influence sur l'équilibre. À contrario les variations des quantités d'eau et d'ions dans le milieu réactionnel liquide ont une influence sur l'équilibre.

Une bouteille d'eau est gazéifiée à l'aide d'une cartouche de CO2. Si l'équilibre de pression est atteint entre la bouteille et la cartouche, la fermeture ou l'ouverture de la vanne entre bouteille et cartouche ne modifie pas la solubilité du gaz dans l'eau.
Le dioxyde de carbone gazeux se dissout dans l'eau liquide en formant l'acide carbonique H2CO3 selon :
Le quotient de réaction s'écrit :

On considère le CO2 gazeux comme un gaz parfait. Son état standard est pris comme étant le gaz parfait pur sous une pression de = 1 bar à la même température que le milieu réactionnel et son activité vaut : , où est la pression totale si le CO2 est pur, ou la pression partielle du CO2 si celui-ci est en présence d'un autre gaz (par exemple d'air). L'eau est le solvant du milieu réactionnel liquide, elle est l'espèce ultra-majoritaire dans ce milieu et peut donc être considérée comme un corps pur. Son état standard est pris comme étant l'eau liquide pure à la même température que le milieu réactionnel et son activité est considérée comme celle d'un corps pur : . Pour le H2CO3, l'état standard considéré est l'espèce à une concentration de 1 mol/l en solution aqueuse à la même température que le milieu réactionnel. Son activité est assimilée à sa concentration : . On retrouve à l'équilibre la loi de Henry de la réaction de dissolution :



![{\displaystyle a_{\rm {H_{2}CO_{3}}}^{\rm {aq}}=\left[{\rm {H_{2}CO_{3}}}\right]/c^{\circ }=\left[{\rm {H_{2}CO_{3}}}\right]}](https://img.franco.wiki/i/de0f2d44945506b8a4ff5e9e6efacba2bf2c8243.svg)
![{\displaystyle K={\left[{\rm {\rm {H_{2}CO_{3}}}}\right] \over P_{\rm {CO_{2}}}}}](https://img.franco.wiki/i/58adb774a1a16e611a836ca115d359d907b2a94f.svg)
Lorsque la saturation est atteinte, l'ajout ou l'extraction de CO2 en phase gaz n'a aucune influence sur la concentration du H2CO3 du moment que la pression n'est pas modifiée. Ainsi, lorsque l'on gazéifie une bouteille d'eau à l'aide d'une cartouche de CO2, la fermeture de la vanne entre la cartouche et la bouteille, lorsque l'équilibre de pression est atteint entre la cartouche et la bouteille, ne modifie pas la concentration en H2CO3 dans la bouteille : en fermant la vanne on retire du ciel gazeux de la bouteille la quantité de gaz correspondant à la cartouche, mais la pression dans la bouteille ne change pas. De même, si l'on rouvre la vanne, on rajoute la quantité de gaz de la cartouche au ciel gazeux de la bouteille, mais, la pression de la bouteille étant inchangée (puisqu'elle est égale à celle de la cartouche), la concentration en H2CO3 ne varie pas.
En revanche, si l'on ajoute une base au milieu aqueux, on favorise la formation de bicarbonate HCO3− et de carbonate CO32− à partir du H2CO3. On extrait donc l'acide carbonique du milieu aqueux, sa concentration baisse, et l'équilibre se rétablit par dissolution du CO2 gazeux. Si au contraire on acidifie le milieu, la concentration en ions diminue, celle de H2CO3 augmente et l'équilibre se rétablit par dégazage de CO2. C'est pourquoi la dissolution du CO2 est bien plus importante en milieu basique qu'en milieu acide ou neutre[71] et du CO2 se dissout pour rétablir l'équilibre. Si l'on évapore de l'eau on concentre le H2CO3 et du CO2 dégaze pour rétablir l'équilibre.
En conclusion, la variation de la quantité de CO2 dans la phase gazeuse hétérogène n'a aucune influence sur l'équilibre à pression constante. À contrario les variations des quantités d'eau et de H2CO3 dans le milieu réactionnel liquide ont une influence sur l'équilibre.
Cas des solutions idéales à pression constante
Ce chapitre détaille l'évolution d'un équilibre chimique dont le milieu réactionnel se comporte comme une solution idéale. Ceci concerne donc aussi bien les solutions idéales liquides ou solides que les mélanges de gaz parfaits.
Condition d'évolution spontanée à pression constante
Supposons un équilibre chimique à pression et à température . La quantité du constituant (réactif, produit, inerte) est modifiée à , la pression et la température restant constantes. On a la variation d'affinité chimique[72],[69] :
![{\displaystyle \left({\partial {\mathcal {A}}^{\text{id}} \over \partial n_{i}}\right)_{P,T,n_{j\neq i}}=-\left[\nu _{i}-x_{i}\Sigma \nu \right]\cdot {RT \over n_{i}}}](https://img.franco.wiki/i/66d91d67027c695353d9b56ef4933a051276c108.svg)
On obtient la relation qui détermine le sens de déplacement de la réaction par modification de la composition :
![{\displaystyle -\left[\nu _{i}-x_{i}\Sigma \nu \right]\,RT\,{\mathrm {d} n_{i} \over n_{i}}\,\mathrm {d} \xi \geq 0}](https://img.franco.wiki/i/90b865592dccbf6f367a4b793f880c8b163a562e.svg)
avec :
- la fraction molaire du constituant dans l'équilibre initial ;
- le coefficient stœchiométrique du constituant ;
- la somme des coefficients stœchiométriques de la réaction.

Les coefficients stœchiométriques sont notés selon la convention stœchiométrique (coefficients des réactifs négatifs).
Dans le cas particulier où , soit , la condition devient :


![{\displaystyle \nu _{i}\,\left[1-x_{i}\right]\,{RT \over 2}\,\left({\mathrm {d} n_{i} \over n_{i}}\right)^{2}\,\mathrm {d} \xi \geq 0}](https://img.franco.wiki/i/d3e63c57847a1847da7baa46bfbd14477e5160fb.svg)
Puisque , ce cas ne peut apparaitre que si .


Dans une solution idéale, y compris les mélanges de gaz parfaits, par définition, le potentiel chimique de tout constituant (réactif, produit, inerte) vaut :

avec :
- la quantité du constituant (réactif, produit, inerte) ;
- la quantité de matière totale du mélange réactionnel (réactifs, produits et inertes) ;
- la fraction molaire du constituant , ;
- le potentiel chimique du constituant dans la solution idéale ;
- le potentiel chimique du constituant pur, aux mêmes , et phase que la solution idéale ;





d'où la dérivée partielle du potentiel chimique dans le cas de la solution idéale :

Avec :

on a pour la solution idéale :

![{\displaystyle \left({\partial {\mathcal {A}}^{\text{id}} \over \partial n_{i}}\right)_{P,T,n_{j\neq i}}=-\sum _{k=1}^{N}\nu _{k}\left({\partial \mu _{k}^{\text{id}} \over \partial n_{i}}\right)_{P,T,n_{j\neq i}}=-RT\,\left[{\nu _{i} \over n_{i}}-{\sum _{k=1}^{N}\nu _{k} \over n}\right]=-RT\,\left[\nu _{i}-x_{i}\Sigma \nu \right]\cdot {1 \over n_{i}}}](https://img.franco.wiki/i/4457bf32dfe6b3522d1977ff19a85909f7fff4db.svg)
Dans le cas particulier où , il est nécessaire d'étudier la dérivée seconde de l'affinité :
![{\displaystyle \left({\partial ^{2}{\mathcal {A}}^{\text{id}} \over {\partial n_{i}}^{2}}\right)_{P,T,n_{j\neq i}}=RT\,\left[{\nu _{i} \over {n_{i}}^{2}}-{\sum _{k=1}^{N}\nu _{k} \over n^{2}}\right]=RT\,\left[\nu _{i}-{x_{i}}^{2}\Sigma \nu \right]\cdot {1 \over {n_{i}}^{2}}}](https://img.franco.wiki/i/4e9af04c2015097921d210d0aecce83d224a12d8.svg)
Avec l'affinité à l'équilibre initial et l'affinité après ajout de constituant en l'absence de réaction, le développement en série de Taylor donne, à pression et température constantes :

![{\displaystyle {\begin{aligned}{\mathcal {A}}^{\text{id}}&\approx {\mathcal {A}}_{0}+\mathrm {d} {\mathcal {A}}^{\text{id}}+{1 \over 2}\,\mathrm {d} ^{2}{\mathcal {A}}^{\text{id}}\approx {\mathcal {A}}_{0}+\left({\partial {\mathcal {A}}^{\text{id}} \over \partial n_{i}}\right)_{P,T,n_{j\neq i}}\,\mathrm {d} n_{i}+{1 \over 2}\,\left({\partial ^{2}{\mathcal {A}}^{\text{id}} \over {\partial n_{i}}^{2}}\right)_{P,T,n_{j\neq i}}\,\left(\mathrm {d} n_{i}\right)^{2}\\&\approx -RT\cdot \left[\nu _{i}-x_{i}\Sigma \nu \right]\,{\mathrm {d} n_{i} \over n_{i}}+{RT \over 2}\cdot \left[\nu _{i}-{x_{i}}^{2}\Sigma \nu \right]\,\left({\mathrm {d} n_{i} \over n_{i}}\right)^{2}\end{aligned}}}](https://img.franco.wiki/i/3948d0480f0cd8b469320836b3d0685e91c2294d.svg)
Dans le cas où , on étudie :

![{\displaystyle {\mathcal {A}}^{\text{id}}\approx \mathrm {d} {\mathcal {A}}^{\text{id}}=-RT\cdot \left[\nu _{i}-x_{i}\Sigma \nu \right]\,{\mathrm {d} n_{i} \over n_{i}}}](https://img.franco.wiki/i/282bdcaee1f58eb85807e852a59d472e5c9922af.svg)
Dans le cas particulier où , soit , on a , on étudie :

![{\displaystyle {\mathcal {A}}^{\text{id}}\approx {1 \over 2}\,\mathrm {d} ^{2}{\mathcal {A}}^{\text{id}}={RT \over 2}\cdot \left[\nu _{i}-{x_{i}}^{2}\Sigma \nu \right]\,\left({\mathrm {d} n_{i} \over n_{i}}\right)^{2}={RT \over 2}\cdot \nu _{i}\,\left[1-{x_{i}}^{2}{\Sigma \nu \over \nu _{i}}\right]\,\left({\mathrm {d} n_{i} \over n_{i}}\right)^{2}={RT \over 2}\cdot \nu _{i}\,\left[1-x_{i}\right]\,\left({\mathrm {d} n_{i} \over n_{i}}\right)^{2}}](https://img.franco.wiki/i/f0b7bf80df594947f4b5d16573124b72646143ea.svg)
La condition d'évolution spontanée après modification de la composition est :

Dans une solution idéale, y compris les mélanges de gaz parfaits, par définition, le potentiel chimique de tout constituant (réactif, produit, inerte) vaut :
avec :
- la quantité du constituant (réactif, produit ou inerte) ;
- la quantité de matière totale dans le mélange ;
- la fraction molaire du constituant ;
- le potentiel chimique du constituant pur dans la même phase que le mélange, sous la pression et à la température .

Pour le constituant dont on modifie la quantité, on a :
![{\displaystyle \left({\partial \mu _{i}^{\text{id}} \over \partial n_{i}}\right)_{P,T,n_{k\neq i}}=RT\,\left({\partial \ln x_{i} \over \partial n_{i}}\right)_{P,T,n_{k\neq i}}=RT\,\left[\left({\partial \ln n_{i} \over \partial n_{i}}\right)_{P,T,n_{k\neq i}}-\left({\partial \ln n \over \partial n_{i}}\right)_{P,T,n_{k\neq i}}\right]=RT\,\left[{1 \over n_{i}}-{1 \over n}\right]}](https://img.franco.wiki/i/4b268ab73f3efd4d58a34527c87697f9985cdab7.svg)
On vérifie la relation, puisque :


Par ailleurs, puisque pour tout constituant (réactif, produit, inerte) , on a également :
![{\displaystyle \left({\partial \mu _{i}^{\text{id}} \over \partial \xi }\right)_{P,T}=RT\,\left({\partial \ln x_{i} \over \partial \xi }\right)_{P,T}=RT\,\left[\left({\partial \ln n_{i} \over \partial \xi }\right)_{P,T}-\left({\partial \ln n \over \partial \xi }\right)_{P,T}\right]=RT\,\left[{\nu _{i} \over n_{i}}-{\Sigma \nu \over n}\right]=\left[\nu _{i}-x_{i}\Sigma \nu \right]{RT \over n_{i}}}](https://img.franco.wiki/i/315e308eaf057efd5fdc468593c35003351660f9.svg)
On vérifie la relation :

Considérons la fraction molaire du constituant (réactif, produit, inerte). Avant ajout du constituant on a :
- la quantité de : ;
- la quantité de matière totale : ;
- la fraction molaire de : .


Dans un premier temps on considère l'ajout d'une quantité en l'absence de réaction ; on a :
- la quantité de : ;
- la variation de la quantité de : ;
- la quantité de matière totale : ;
- la variation de la quantité de matière totale : .




Par conséquent la variation de la fraction molaire par ajout de constituant seul vaut :

On a :
![{\displaystyle \mathrm {d} x_{i}^{\prime }\cdot \mathrm {d} n_{i}=\left[{1 \over n_{i}}-{1 \over n}\right]\cdot x_{i}\cdot \left(\mathrm {d} n_{i}\right)^{2}}](https://img.franco.wiki/i/426a4291d7fcdd70bdb75ee1437a3eced9decf14.svg)
Ainsi, puisque :

Puis l'on considère l'ajout de constituant et le déplacement dû à la réaction combinés ; on a :
- la quantité de : ;
- la variation de la quantité de : ;
- la quantité de matière totale : ;
- la variation de la quantité de matière totale : .




Par conséquent la variation de la fraction molaire par ajout de constituant et déplacement de réaction combinés vaut :

On a :
![{\displaystyle {\mathrm {d} x_{i}^{\prime \prime } \over x_{i}}=\left[{1 \over n_{i}}-{1 \over n}\right]\cdot \mathrm {d} n_{i}+\left[{\nu _{i} \over n_{i}}-{\Sigma \nu \over n}\right]\cdot \mathrm {d} \xi }](https://img.franco.wiki/i/1479c50d09ad6fa1b44b4098619a21ad8f0872a4.svg)
![{\displaystyle {\mathrm {d} x_{i}^{\prime \prime }-\mathrm {d} x_{i}^{\prime } \over x_{i}}=\left[{\nu _{i} \over n_{i}}-{\Sigma \nu \over n}\right]\cdot \mathrm {d} \xi }](https://img.franco.wiki/i/a5fe39252b79a8abf3c9beb98a163b0449afd570.svg)
On a par la condition d'évolution spontanée de la réaction :
![{\displaystyle \left({\partial {\mathcal {A}}^{\text{id}} \over \partial n_{i}}\right)_{P,T,n_{k\neq i}}\cdot \mathrm {d} n_{i}\cdot \mathrm {d} \xi =-RT\,\left[{\nu _{i} \over n_{i}}-{\Sigma \nu \over n}\right]\cdot \mathrm {d} n_{i}\cdot \mathrm {d} \xi =-RT\,{\mathrm {d} x_{i}^{\prime \prime }-\mathrm {d} x_{i}^{\prime } \over x_{i}}\cdot \mathrm {d} n_{i}\geq 0}](https://img.franco.wiki/i/2547af02a082209d39329d2b715b2dd4c0933949.svg)
et par conséquent :

Ainsi, si l'on considère un ajout de constituant, soit , alors :


Par ajout du constituant en l'absence de réaction, la fraction molaire de ce constituant augmente et devient . Par ajout du constituant et déplacement de réaction combinés, la fraction obtenue finalement est égale à . La réaction s'oppose donc à l'augmentation de la fraction molaire du constituant ajouté[49].


En ce qui concerne le potentiel chimique du constituant, on a :
- le potentiel chimique à l'équilibre initial : ;
- le potentiel chimique après ajout du constituant en l'absence de réaction : ;
- le potentiel chimique après ajout du constituant et réaction : .



On vérifie que :


La réaction s'est donc opposée à l'augmentation du potentiel chimique du constituant ajouté.
Pour les solutions idéales, y compris les mélanges de gaz parfaits, dont on modifie la composition à pression et température constantes, le principe de Le Chatelier est respecté si l'on considère la fraction molaire du constituant dont on modifie la quantité : si l'on augmente la fraction molaire d'un constituant, la réaction tend à la diminuer[51],[49].
Ajout ou extraction d'un réactif ou d'un produit à pression constante
Dans le cas de l'ajout ou de l'extraction d'un réactif ou d'un produit, c'est-à-dire d'un constituant intervenant dans la réaction, , à pression et température constantes il vient six cas de figure[72],[69] :

- :
- si l'on ajoute le constituant : , d'où , la réaction progresse ;
- si l'on extrait le constituant : , d'où , la réaction régresse ;
- :
- si l'on ajoute le constituant : , d'où , la réaction régresse ;
- si l'on extrait le constituant : , d'où , la réaction progresse ;
- :
- si est un réactif : , d'où , la réaction régresse que l'on ajoute ou extraie ;
- si est un produit : , d'où , la réaction progresse que l'on ajoute ou extraie .




Ceci se résume en termes de fraction molaire du constituant ajouté :
| L'ajout d'un réactif ou d'un produit à pression et température constantes déplace l'équilibre dans le sens qui diminue la fraction molaire de ce constituant[51],[49]. | Inversement, l'extraction d'un réactif ou d'un produit à pression et température constantes déplace l'équilibre dans le sens qui augmente la fraction molaire de ce constituant. |
Si le principe de Le Chatelier est toujours respecté : l'ajout d'un réactif () ou l'extraction d'un produit () fait progresser la réaction ; à contrario l'extraction d'un réactif ou l'ajout d'un produit fait régresser la réaction.
Si , pour tout constituant (réactif ou produit) pour lequel , le sens de déplacement de la réaction dépend du positionnement de par rapport à . En conséquence, selon la composition du mélange dans l'équilibre initial, l'ajout d'un constituant pourra déplacer l'équilibre dans un sens ou dans l'autre. Le principe de Le Chatelier peut ne pas être respecté au regard de la quantité du constituant ajouté. Par exemple, l'ajout d'un réactif peut déplacer l'équilibre dans le sens de la régression de la réaction (production du réactif ajouté), et non de sa progression (consommation du réactif ajouté) comme le voudrait le principe de modération. La réaction s'oppose néanmoins à l'augmentation de la fraction molaire de ce constituant. Cette fraction a pour expression . La réaction modifie à la fois la quantité du constituant et la quantité de matière totale , celles-ci pouvant varier dans le même sens ou en sens opposés. La réaction se déplace dans le sens qui diminue la fraction , quitte à ce que la quantité augmente du moment que la quantité de matière totale augmente suffisamment pour que diminue. Le déplacement d'équilibre respecte le principe de Le Chatelier au regard de la fraction molaire et non de la quantité du constituant modifié.




Cet exemple présente un cas de violation du principe de modération. Reprenons la réaction de synthèse de l'ammoniac dans le procédé Haber-Bosch :
La somme des coefficients stœchiométriques vaut : .

Pour l'ammoniac, produit dont le coefficient stœchiométrique vaut : , nous avons , quelle que soit la valeur de la fraction molaire initiale d'ammoniac , nous avons , d'où :



- lorsque l'on ajoute de l'ammoniac la réaction régresse ;
- lorsque l'on extrait de l'ammoniac la réaction progresse ;
- le principe de Le Chatelier est respecté.
Pour l'hydrogène, réactif dont le coefficient stœchiométrique vaut : , nous avons , quelle que soit la valeur de la fraction molaire initiale d'hydrogène , nous avons , d'où :



- lorsque l'on ajoute de l'hydrogène la réaction progresse ;
- lorsque l'on extrait de l'hydrogène la réaction régresse ;
- le principe de Le Chatelier est respecté.
Pour l'azote, réactif dont le coefficient stœchiométrique vaut : , nous avons :


- si la fraction molaire initiale d'azote est , alors :
- lorsque l'on ajoute de l'azote la réaction progresse ;
- lorsque l'on extrait de l'azote la réaction régresse ;
- le principe de Le Chatelier est respecté ;
- si la fraction molaire initiale d'azote est , alors :
- lorsque l'on ajoute de l'azote la réaction régresse ;
- lorsque l'on extrait de l'azote la réaction progresse ;
- le principe de Le Chatelier n'est pas respecté ;
- si la fraction molaire initiale d'azote est , alors :
- que l'on ajoute ou extraie de l'azote la réaction régresse ;
- le principe de Le Chatelier n'est pas respecté.






Il est donc contre-productif de travailler avec un trop grand excès d'azote dans le procédé Haber-Bosch de synthèse de l'ammoniac.
Un autre exemple de violation du principe de Le Chatelier est donné par la synthèse en phase gaz du méthanol CH3OH à partir de l'hydrogène H2 et du monoxyde de carbone CO :

Si la fraction molaire initiale de CO est et que l'on ajoute du CO, la réaction se déplace dans le sens de la destruction du méthanol en monoxyde de carbone, donc dans le sens de l'apparition du réactif ajouté.

L'effet d'ion commun est un exemple d'application du principe de modération. La dissolution du chlorure de sodium (NaCl), ou sel de table, est donnée par la réaction :
La solubilité du chlorure de sodium est donnée par le produit de solubilité :
![{\displaystyle K_{\text{NaCl}}={\rm {\left[Na^{+}\right]\left[Cl^{-}\right]}}}](https://img.franco.wiki/i/74db09442d1d0e52c788c9df32cae612e644bdb6.svg)
Si l'on ajoute un autre sel contenant un ion commun avec le chlorure de sodium, par exemple le sulfate de sodium, qui lui-même se dissout selon la réaction :

on augmente la concentration des ions Na+. En conséquence, l'équilibre du chlorure de sodium se déplace dans le sens de la consommation de ce cation, c'est-à-dire de la précipitation du NaCl, de droite à gauche. De même, la dissolution de chlorure d'hydrogène HCl gazeux selon :

augmente la concentration en Cl−. En conséquence, l'équilibre du chlorure de sodium se déplace dans le sens de la consommation de cet anion, de droite à gauche.
Au contraire, si l'on ajoute de l'acide sulfurique H2SO4 à la solution saline, celui-ci se dissout selon :

La concentration en ions SO2−
4 augmente et fait précipiter le sulfate de sodium Na2SO4 selon la réaction vue plus haut. La concentration en Na+ diminue, en conséquence l'équilibre du NaCl se déplace dans le sens de la production de ce cation, celui de la dissolution du sel, de gauche à droite.
Ajout ou extraction d'un inerte à pression constante
Dans le cas de l'ajout ou de l'extraction d'un constituant inerte, c'est-à-dire d'un constituant qui n'intervient pas dans la réaction, , à pression et température constantes, on a la variation d'affinité chimique[72],[75] :


On obtient la relation qui détermine le sens de déplacement de la réaction par modification de la composition :

avec la quantité de matière totale du mélange réactionnel (réactifs, produits et inertes).
Il vient cinq cas de figure[72],[75] :
- , la réaction ne modifie pas la quantité totale de constituants (réactifs et produits, hors inertes) ; on a, pour tout entier naturel non nul, ; l'affinité chimique n'est pas modifiée par la modification de la quantité d'inerte, elle reste donc nulle : l'équilibre est neutre, insensible à la quantité d'inerte, et ne se déplace pas ;
- , le nombre total de constituants (réactifs et produits, hors inertes) diminue lorsque l'équilibre se déplace de la gauche vers la droite :
- si l'on ajoute de l'inerte : , d'où , la réaction régresse ;
- si l'on extrait de l'inerte : , d'où , la réaction progresse ;
- , le nombre total de constituants (réactifs et produits, hors inertes) augmente lorsque l'équilibre se déplace de la gauche vers la droite :
- si l'on ajoute de l'inerte : , d'où , la réaction progresse ;
- si l'on extrait de l'inerte : , d'où , la réaction régresse.

Pour résumer :
| L'ajout d'un inerte à pression et température constantes déplace l'équilibre dans le sens qui augmente la quantité totale de constituants (réactifs et produits) dans le mélange[72],[75]. | Inversement, l'extraction d'un inerte à pression et température constantes déplace l'équilibre dans le sens qui diminue la quantité totale de constituants (réactifs et produits) dans le mélange. |
Ce résultat peut sembler paradoxal : on pourrait s'attendre à ce que l'ajout d'un inerte déplace l'équilibre dans le sens d'une diminution de la quantité globale de réactifs et de produits, et non à une augmentation. Or la réaction, dans les conditions de pression et température constantes, doit tendre à diminuer la fraction molaire du constituant ajouté. Cette fraction a pour expression . La réaction n'ayant aucun impact sur la quantité d'un inerte, par définition, elle ne peut diminuer sa fraction molaire qu'en faisant augmenter la quantité de matière totale . Dans le cas particulier où , la réaction ne modifie pas la quantité de matière totale : l'ajout ou l'extraction d'inerte est sans effet sur l'équilibre.
La réaction de synthèse de l'iodure d'hydrogène à partir d'hydrogène et d'iode s'écrit :
La somme des coefficients stœchiométriques est donc de = –1 –1 +2 = 0. Cette réaction est insensible à la présence d'inertes à pression et température constantes.
Reprenons la réaction de synthèse de l'ammoniac dans le procédé Haber-Bosch :
La somme des coefficients stœchiométriques vaut : = –2. Puisque cette somme est négative, l'ajout d'un inerte à pression et température constantes déplace l'équilibre dans le sens de la production d'azote et d'hydrogène. Pour un bon rendement de la réaction, il vaut mieux éviter l'introduction d'inertes dans le mélange réactionnel.
Un électrolyte faible totalement dissout dans l'eau se dissocie partiellement selon la réaction :


avec le cation et l'anion associés, et le nombre de charges de ces ions.



Dans cette réaction l'eau est le solvant et a un rôle d'inerte. En conséquence, lorsque l'on augmente la quantité d'eau dans le milieu réactionnel à pression et température constantes, la réaction se déplace dans le sens de la production du cation et de l'anion. Il s'agit de la loi de dilution d'Ostwald : la dilution d'un électrolyte faible induit le déplacement de la réaction dans le sens de la dissociation de l'électrolyte.
Récapitulatif pour la modification de composition à pression constante
Le tableau suivant récapitule les divers cas de figure de déplacement de l'équilibre chimique en solution idéale, y compris les mélanges de gaz parfaits, par modification de la composition à pression et température constantes. On note :
- la fraction molaire du constituant dans le mélange réactionnel à l'équilibre avant modification de la composition ;
- le coefficient stœchiométrique du constituant ;
- la somme des coefficients stœchiométriques de la réaction.
Les coefficients stœchiométriques sont notés selon la convention stœchiométrique (coefficient d'un réactif négatif).
| Critères stœchiométriques | Opération | Déplacement | |
|---|---|---|---|
réactif | ajout | progression | |
| extraction | régression | ||
| ajout | régression | ||
| extraction | régression | ||
| ajout | régression | ||
| extraction | progression | ||
produit | ajout | progression | |
| extraction | régression | ||
| ajout | progression | ||
| extraction | progression | ||
| ajout | régression | ||
| extraction | progression | ||
inerte | ajout | régression | |
| extraction | progression | ||
| ajout | sans effet | ||
| extraction | sans effet | ||
| ajout | progression | ||
| extraction | régression | ||
De façon générale, l'ajout d'un constituant (réactif, produit, inerte) en solution idéale, y compris dans les mélanges de gaz parfaits, à pression et température constantes, déplace l'équilibre dans le sens qui diminue la fraction molaire de ce constituant[51],[49].
Cas des gaz parfaits à volume constant
Ce chapitre détaille l'évolution d'un équilibre chimique dont le milieu réactionnel est un mélange de gaz parfaits. Concernant les phases condensées (liquides en particulier), il est en pratique impossible d'ajouter ou d'extraire un composant sans faire varier le volume du milieu réactionnel.
Condition d'évolution spontanée à volume constant
Supposons un équilibre chimique au volume et à température . La quantité du constituant (réactif, produit, inerte) est modifiée à , le volume et la température restant constants. On a la variation d'affinité chimique[74] :

On obtient la relation qui détermine le sens de déplacement de la réaction par modification de la composition[74] :

avec le coefficient stœchiométrique du constituant noté selon la convention stœchiométrique (coefficients des réactifs négatifs).
Dans un mélange de gaz parfaits, par définition de la fugacité, le potentiel chimique de tout constituant (réactif, produit, inerte) vaut :

avec :
- la quantité du constituant (réactif, produit, inerte) ;
- la quantité de matière totale du mélange réactionnel (réactifs, produits et inertes) ;
- la pression ;
- une pression de référence constante, en général 1 bar ;
- la fraction molaire du constituant , ;
- le potentiel chimique du constituant dans le mélange de gaz parfaits ;
- le potentiel chimique du constituant à l'état de gaz parfait pur, sous la pression et à la température .


Selon la loi des gaz parfaits :


d'où :


On a pour un mélange de gaz parfaits :


Dans un mélange de gaz parfaits, voir démonstration ci-dessus, le potentiel chimique de tout constituant (réactif, produit, inerte) vaut :

avec :
- la quantité du constituant (réactif, produit ou inerte) ;
- une pression de référence constante, en général 1 bar ;
- le potentiel chimique du constituant à l'état de gaz parfait pur, sous la pression et à la température .
Pour le constituant dont on modifie la quantité, on a :

On vérifie la relation :

Par ailleurs, puisque pour tout constituant (réactif, produit, inerte) , on a également :

On vérifie la relation :

Considérons la quantité du constituant (réactif, produit, inerte). Avant ajout du constituant on a la quantité de : .
Dans un premier temps on considère l'ajout d'une quantité en l'absence de réaction. On a :
- la quantité de : ;
- la variation de la quantité de : .
Puis l'on considère l'ajout de constituant et le déplacement dû à la réaction combinés. On a :
- la quantité de : ;
- la variation de la quantité de : .
On a :

On a par la condition d'évolution spontanée de la réaction :
![{\displaystyle \left({\partial {\mathcal {A}} \over \partial n_{i}}\right)_{V,T,n_{k\neq i}}\cdot \mathrm {d} n_{i}\cdot \mathrm {d} \xi =-\nu _{i}{RT \over n_{i}}\cdot \mathrm {d} n_{i}\cdot \mathrm {d} \xi =-\left[\mathrm {d} n_{i}^{\prime \prime }-\mathrm {d} n_{i}^{\prime }\right]\cdot \mathrm {d} n_{i}\geq 0}](https://img.franco.wiki/i/a6499dfbbe4cf680fa46bcd5577d8732da8c3e96.svg)
et par conséquent :

Ainsi, si l'on considère un ajout de constituant, soit , alors .

Par ajout du constituant en l'absence de réaction, la quantité de ce constituant augmente. Par ajout du constituant et déplacement de réaction combinés, la quantité peut augmenter ou diminuer, mais finalement . La réaction s'oppose donc à l'augmentation de la quantité du constituant ajouté.

En ce qui concerne le potentiel chimique du constituant, on a :
- le potentiel chimique à l'équilibre initial : ;
- le potentiel chimique après ajout du constituant en l'absence de réaction : ;
- le potentiel chimique après ajout du constituant et réaction : .



On vérifie que :


La réaction s'est donc opposée à l'augmentation du potentiel chimique du constituant ajouté.
Pour les mélanges de gaz parfaits dont on modifie la composition à volume et température constants, le principe de Le Chatelier est respecté si l'on considère la quantité du constituant que l'on modifie : si l'on augmente la quantité d'un constituant, la réaction tend à la diminuer.
Ajout ou extraction d'un réactif ou d'un produit à volume constant
Dans le cas de l'ajout ou de l'extraction d'un réactif ou d'un produit, c'est-à-dire d'un constituant intervenant dans la réaction, , à volume et température constants il vient quatre cas de figure[74] :
- :
- si l'on ajoute le réactif : , d'où , la réaction progresse ;
- si l'on extrait le réactif : , d'où , la réaction régresse ;
- :
- si l'on ajoute le produit : , d'où , la réaction régresse ;
- si l'on extrait le produit : , d'où , la réaction progresse.
Pour résumer :
| À volume et température constants l'ajout d'un réactif ou d'un produit déplace l'équilibre dans le sens de la consommation de ce constituant[74]. | Inversement, à volume et température constants l'extraction d'un réactif ou d'un produit déplace l'équilibre dans le sens de la production de ce constituant. |
Le principe de Le Chatelier est toujours respecté.
Ajout ou extraction d'un inerte à volume constant
Dans le cas d'un constituant inerte, c'est-à-dire d'un constituant qui n'intervient pas dans la réaction, , à volume et température constants, on a la variation d'affinité chimique[67] :

On a également, pour tout entier naturel non nul :

L'affinité chimique n'est pas modifiée par la modification de la quantité d'inerte, elle reste donc nulle ; l'équilibre est insensible à la quantité d'inerte et ne se déplace pas[67] :
| À volume et température constants l'ajout ou l'extraction d'un inerte est sans effet sur l'équilibre[67]. |
Ce résultat peut sembler paradoxal : on pourrait s'attendre à ce que l'ajout d'un inerte déplace l'équilibre dans le sens d'une diminution de la quantité globale de réactifs et de produits. Or la réaction, dans les conditions de volume et température constants, doit tendre à diminuer la quantité du constituant ajouté. La réaction n'ayant aucun impact sur la quantité d'un inerte, par définition, la modification de celle-ci n'a aucune influence sur l'équilibre.
Récapitulatif pour la modification de composition à volume constant
Le tableau suivant récapitule les divers cas de figure de déplacement de l'équilibre chimique dans un mélange de gaz parfaits par modification de la composition à volume et température constants.
| Constituant | Opération | Déplacement |
|---|---|---|
réactif | ajout | progression |
| extraction | régression | |
produit | ajout | régression |
| extraction | progression | |
inerte | ajout | sans effet |
| extraction | sans effet |
De façon générale, l'ajout d'un constituant dans les mélanges de gaz parfaits, à volume et température constants, déplace l'équilibre dans le sens qui diminue la quantité de ce constituant. La réaction n'ayant aucun effet sur la quantité d'un inerte, cette modification est sans effet.
Emploi d'un catalyseur
Un catalyseur augmente la vitesse des réactions en abaissant leur énergie d'activation. Un catalyseur permet d'atteindre plus vite l'équilibre.
Les concentrations finales sont identiques avec ou sans catalyseur. Un catalyseur ne permet pas de déplacer un équilibre.
Un catalyseur augmente la vitesse des réactions en abaissant leur énergie d'activation ; il permet donc d'atteindre plus vite l'équilibre[77],[78]. Un catalyseur agit cependant de façon identique sur la réaction directe et la réaction inverse. Autrement dit, si l'énergie d'activation de la réaction directe varie de par l'effet du catalyseur, devenant , alors l'énergie d'activation de la réaction inverse devient . L'enthalpie standard de réaction (voir le paragraphe Loi d'Arrhenius) n'est donc pas modifiée, puisque avant ajout du catalyseur on a :




et après ajout du catalyseur on a :
![{\displaystyle \left[E_{\rightarrow }^{\ddagger }+\Delta E^{\ddagger }\right]-\left[E_{\leftarrow }^{\ddagger }+\Delta E^{\ddagger }\right]=E_{\rightarrow }^{\ddagger }-E_{\leftarrow }^{\ddagger }=\Delta _{\text{r}}H^{\circ }}](https://img.franco.wiki/i/fa6140c5edd8208f361a20af4be79d3186074a75.svg)
L'entropie standard de réaction n'étant pas non plus modifiée par le catalyseur, l'enthalpie libre standard de réaction et par conséquent la constante d'équilibre ne sont donc également pas modifiées par le catalyseur :

En conclusion l'emploi d'un catalyseur ne permet pas de déplacer un équilibre. Il ne permet pas non plus de rendre possible une réaction thermodynamiquement impossible[77].
| Un catalyseur permet d'atteindre plus rapidement l'équilibre, mais il ne permet pas de le déplacer[77],[78]. |
Le raisonnement est le même pour un inhibiteur qui ralentit la réaction en augmentant son énergie d'activation (soit ) : l'équilibre est atteint plus lentement, mais il n'est pas déplacé.

Équilibres chimiques simultanés
Généralités
Stœchiométrie, avancements de réaction
Soit un milieu réactionnel siège de réactions simultanées impliquant constituants , on note les diverses réactions selon[15],[79] :






On note :
- l'indice des réactions, ;
- l'indice des constituants, ;
- le coefficient stœchiométrique du constituant dans la réaction , tel que :
- pour un réactif ;
- pour un produit ;
- si le constituant n'intervient pas dans la réaction .
![{\displaystyle i\in \left[1,\cdots ,M\right]}](https://img.franco.wiki/i/85ab0f4f2f429ba69aa18e1af3a11229160307a6.svg)
![{\displaystyle j\in \left[1,\cdots ,N\right]}](https://img.franco.wiki/i/a7dbde8c8aed36bad5c74c1493692b16fda9464d.svg)




Soit la variation de quantité du constituant due à la réaction . On a si , c'est-à-dire si le constituant n'intervient pas dans la réaction . Pour chacun des constituants, on a la variation globale de quantité dans le mélange réactionnel :




Pour chacune des réactions, on définit un avancement de réaction tel que pour chacun des constituants :


On a donc pour tout constituant , réactif, produit ou inerte[79] :

ou, en intégrant entre l'instant initial et un instant quelconque[15],[79] :

avec :
- la quantité du constituant à l'instant initial ;
- la quantité du constituant à l'instant ;
- l'avancement de réaction à l'instant ; rappelons qu'à l'instant initial, par définition, .


Cinq constituants () sont considérés :

- le méthane () ;
- l'eau () ;
- l'hydrogène () ;
- le monoxyde de carbone () ;
- le dioxyde de carbone () ;








impliqués dans deux réactions simultanées () en phase gaz :

- réaction : ;
- réaction : (réaction du gaz à l'eau).




Les coefficients stœchiométriques valent en conséquence :

On suppose un mélange initial composé exclusivement de méthane et d'eau ; les quantités initiales d'hydrogène, de monoxyde et de dioxyde de carbone sont nulles : . Les quantités des divers constituants évoluent selon :


La vitesse de la réaction est donnée par la dérivée de l'avancement de cette réaction par rapport au temps [16] :

En conséquence pour chaque constituant la quantité varie par rapport au temps selon[16],[80] :

Enthalpies libres de réaction
Le potentiel chimique de chacun des constituants est défini sur l'ensemble du mélange réactionnel. La variation à pression et température constantes de l'enthalpie libre globale du système réactionnel vaut alors[81],[82] :
![{\displaystyle \mathrm {d} G=\sum _{j=1}^{N}\mu _{j}\,\mathrm {d} n_{j}=\sum _{j=1}^{N}\mu _{j}\left[\sum _{i=1}^{M}\nu _{i,j}\,\mathrm {d} \xi _{i}\right]=\sum _{i=1}^{M}\left[\sum _{j=1}^{N}\nu _{i,j}\mu _{j}\right]\,\mathrm {d} \xi _{i}=\sum _{i=1}^{M}\Delta _{\text{r}}G_{i}\,\mathrm {d} \xi _{i}}](https://img.franco.wiki/i/9496ac9f4b77a091f794ad99c017d6e24d96baf8.svg)
avec l'enthalpie libre de réaction de la réaction . On a par conséquent :


Les potentiels chimiques sont des fonctions de l'ensemble des quantités des constituants du mélange réactionnel : . Chacune des enthalpies libres de réaction est donc fonction de l'ensemble des avancements de réaction :


Constantes d'équilibre, quotients de réaction
Pour chacun des constituants, on choisit un état standard unique, valable pour l'ensemble des réactions. Le potentiel chimique du constituant dans le mélange réactionnel est lié au potentiel du constituant dans son état standard par une activité chimique telle que :




L'activité du constituant est donc également unique et valable pour l'ensemble des réactions du mélange réactionnel. Pour chacune des réactions simultanées, on peut définir une constante d'équilibre et un quotient de réaction :





Ainsi pour toute réaction peut-on écrire :

Évolution du système
Condition d'évolution spontanée, couplage chimique
Le deuxième principe de la thermodynamique induit que, à pression et température constantes, l'enthalpie libre globale du système réactionnel ne peut que décroître, soit[14],[82] :
| Condition d'évolution spontanée de tout système chimique à et constantes : |

Ceci implique que, contrairement au cas d'une réaction seule, une réaction (ou plusieurs réactions) du système réactionnel peut évoluer selon , c'est-à-dire avec et de même signe, du moment que les autres réactions compensent cette augmentation et que décroît. Autrement dit, cette réaction seule diminue l'entropie, mais les autres réactions l'augmentent suffisamment pour que le bilan entropique global du système réactionnel soit positif : le deuxième principe de la thermodynamique est donc respecté. Ainsi, une réaction qui, seule, ne pourrait être spontanée peut être provoquée dans un milieu réactionnel siège de plusieurs réactions dans les mêmes conditions, ce phénomène est appelé couplage chimique[14].



Une réaction exergonique est une réaction qui dégage de l'énergie en progressant : à pression et température constantes la variation d'enthalpie libre engendrée est négative, soit avec et ; elle est spontanée. Une réaction endergonique est une réaction qui a besoin d'un apport d'énergie pour progresser et ne peut donc être spontanée : à pression et température constantes la variation d'enthalpie libre engendrée est positive, soit avec et ; c'est la réaction inverse () qui est spontanée. Un couplage chimique nécessite deux conditions[83] :


- 1. deux réactions chimiques dans lesquelles un produit de l'une est un réactif de l'autre ; par exemple :
- réaction 1 : ;
- réaction 2 : ;
- 2. que l'une des réactions soit fortement exergonique, soit , et l'autre endergonique, soit , la combinaison des deux réactions étant une réaction exergonique : .





La progression de la réaction 1 est spontanée car avec ; celle de la réaction 2 ne l'est pas car avec . Le couplage chimique permet la progression de la réaction 2 : on peut obtenir avec si . La réaction 2 qui n'est pas spontanée peut ainsi être provoquée.






Exemple du couplage de la phosphorylation du glucose, endergonique, et de l'hydrolyse de l'ATP, exergonique.
Le métabolisme, ensemble des réactions chimiques qui se déroulent à l'intérieur d'un être vivant, utilise le couplage chimique. Le catabolisme regroupe l'ensemble des réactions de dégradation des molécules ; ces réactions sont toutes exergoniques. L'anabolisme regroupe l'ensemble des réactions de synthèse des molécules ; ces réactions sont toutes endergoniques. Les réactions du catabolisme rendent possibles les réactions de l'anabolisme[83] - [85].
L'une des réactions exergoniques les plus courantes du métabolisme est la réaction d'hydrolyse de l'adénosine triphosphate (ATP) produisant de l'adénosine diphosphate (ADP) et du phosphate inorganique (Pi) :

Elle est fortement exergonique, son enthalpie libre standard de réaction à pH neutre (pH = 7) est de = −30,5 kJ/mol[alpha 12]. Sa réaction inverse est la phosphorylation de l'ADP, endergonique.

La réaction de phosphorylation du glucose produisant du glucose-6-phosphate est endergonique, elle s'écrit :

Son enthalpie libre standard de réaction est de = 13,8 kJ/mol. Le couplage chimique avec l'hydrolyse de l'ATP, catalysé par l'hexokinase (dans le muscle) ou la glucokinase (dans le foie) en présence de cations magnésium Mg2+ comme cofacteur, donne une réaction exergonique :

Cette réaction a une enthalpie libre standard de réaction de = -30,5 + 13,8 = -16,7 kJ/mol. La glycolyse, la glycogénogénèse, la voie d'Entner-Doudoroff et la voie des pentoses phosphates sont des voies métaboliques débutant par cette réaction.
La réaction exergonique d'hydrolyse du phosphoénolpyruvate produit du pyruvate :

Cette réaction a une enthalpie libre standard de réaction de = −61,9 kJ/mol. Le couplage avec la phosphorylation de l'ADP, catalysé par la pyruvate kinase en présence d'ions magnésium Mg2+, donne une réaction exergonique :

Cette réaction a une enthalpie libre standard de réaction de = -61,9 + 30,5 = −31,4 kJ/mol. Elle permet la régénération de l'ATP en fin de glycolyse.
Condition d'équilibre
L'équilibre correspond au minimum de la fonction , soit :

Ceci implique que l'enthalpie libre de réaction de chacune des réactions est nulle. On obtient ainsi un ensemble de équations à inconnues (les avancements )[14],[82] :
- pour tout

À l'équilibre, on a donc pour chacune des réactions une loi d'action de masse :

On note à l'aide de l'opérateur nabla le gradient de la fonction :

Il s'agit d'un ensemble de fonctions des avancements de réaction . L'équilibre est donc atteint lorsque le gradient est nul :
| Condition d'équilibre : |

Condition de stabilité
Comme pour une réaction seule, la solution mathématique de pour un ensemble de réactions simultanées n'est pas nécessairement unique. Il convient donc également, lors d'une simulation numérique d'un système réactionnel à réactions simultanées, de vérifier qu'une solution obtenue en annulant les enthalpies libres de réaction est bien un minimum stable de la fonction globale du système réactionnel. Pour cela on définit la matrice hessienne de la fonction à pression et température constantes :


Cette matrice est carrée (de dimension ) et, en vertu du théorème de Schwarz, symétrique (pour deux réactions et quelconques : ). D'autre part, pour tous et :



La dérivée se fait à température constante, et les constantes d'équilibre ne dépendent que de la température, d'où :


Au point d'équilibre tel que , pour tout vecteur de variations élémentaires :

avec son vecteur transposé, on pose la différentielle d'ordre 2 de l'enthalpie libre à pression et température constantes :



L'équilibre est stable si et seulement si a atteint un minimum. À partir de cet équilibre ne peut qu'augmenter, l'enthalpie libre est donc convexe à pression et température constantes. La condition de stabilité des équilibres chimiques simultanés est en conséquence vérifiée si et seulement si la matrice hessienne de est définie positive pour la solution de , ce qui se traduit par :
| Condition de stabilité des équilibres simultanés
À l'équilibre la matrice hessienne est définie positive, soit . |

Si la matrice est définie négative au point d'équilibre, soit , alors est concave en ce point et a atteint un maximum : l'équilibre est instable. Si la matrice n'est pas définie, soit , l'équilibre peut être métastable : il est nécessaire d'étudier les différentielles d'ordre supérieur. Comme pour la réaction seule, le système peut avoir atteint un minimum local, il est nécessaire de rechercher le minimum global de la fonction .


Plutôt que de résoudre d'abord le système de équations à inconnues et de tester à postériori la stabilité des diverses solutions, il est préférable de rechercher la meilleure solution directement à l'aide d'un algorithme de minimisation sous contrainte (par exemple la méthode des multiplicateurs de Lagrange) appliqué à la fonction globale, les contraintes étant que la quantité de chacun des constituants doit rester positive et que la masse globale du système réactionnel est conservée.
Notes et références
Notes
- ↑ Le terme de réaction réversible peut porter à confusion, il ne s'agit pas en effet d'une transformation réversible, ne créant pas d'entropie. De même, le terme de réaction partielle ou incomplète peut s'appliquer à une réaction totale dont les réactifs n'ont pas été introduits en stœchiométrie, par exemple une combustion en défaut d'air. Il est donc préférable d'employer le terme de réaction inversible, cf. portail Dunod de la chimie organique.
- ↑ Raymond Defay (1897-1987), physicien et chimiste belge, professeur à l'Université Libre de Bruxelles, cf. IdRef.
- ↑ Voir l'article Entropie d'un corps pur.
- ↑ Berthelot énonça son principe, fruit d'un immense travail d'expérimentation et de collecte de données, en 1879. Il espérait le hisser au rang des grandes lois universelles, au même titre que la loi de la gravité de Newton. En 1884, à peine cinq ans plus tard, la thèse soutenue par Duhem, qui introduisait en Europe le formalisme mathématique de l'Américain Gibbs, et notamment la fonction enthalpie libre , annihila tous les espoirs de Berthelot. La querelle qui s'ensuivit entre les deux hommes, mêlant science et politique, ruina la carrière de Duhem, cf. Vangelis Antzoulatos, Laboratoire SCité, Université Lille 1, « Prévoir les réactions chimiques : un problème controversé au XIXe siècle » [PDF] (consulté le ).
- ↑ Une réaction spontanée, dans laquelle décroît, est appelée réaction exergonique. À contrario une réaction provoquée, dans laquelle croît, est appelée réaction endergonique.
- ↑ Un équilibre instable est défini par des vitesses de réaction extrêmement faibles, conduisant à une variation de l'avancement de réaction quasi nulle, non mesurable. Ceci est exploité lors des opérations de trempe en métallurgie ou en verrerie. Le matériau est chauffé puis brutalement refroidi. L'état ainsi obtenu est un état hors équilibre, différent de celui obtenu par un refroidissement lent. Le matériau acquiert de cette façon des propriétés de résistance différentes du matériau obtenu ordinairement. En chimie la trempe (refroidissement brutal d'un milieu réactionnel) permet de ralentir les réactions et ainsi de figer la composition du milieu réactionnel hors équilibre.
- ↑ Un équilibre métastable est un équilibre dans lequel les réactions n'ont pas lieu faute d'un facteur déclenchant ; par exemple un mélange d'hydrogène et d'oxygène est métastable, il peut être conservé dans le temps sans variation de composition, mais une simple étincelle déclenche une réaction violente produisant de l'eau. Ce terme s'emploie également dans l'état de surfusion de l'eau.
- ↑ Par exemple, pour la fonction l'équilibre se trouve au point tel que , soit à . La première dérivée non nulle au point est d'ordre 5 : , de même que si , alors . Dans les deux cas l'équilibre est métastable.
- ↑ Par exemple, pour la fonction l'équilibre se trouve au point tel que , soit à . La première dérivée non nulle au point est d'ordre 4 : , l'équilibre est stable. Au contraire, si , alors , l'équilibre est instable.
- ↑ Par exemple, pour la fonction les extrémums se trouvent en tel que , soit , et . On a , d'où : est un minimum, est un maximum, est un minimum. Par conséquent est le minimum global car est un minimum local.
- ↑ Voir l'article Équilibre thermodynamique : les potentiels thermodynamiques sont concaves par rapport à leurs variables intensives, soit si est intensive, et convexes par rapport à leurs variables extensives, soit si est extensive.
- ↑ En biochimie les conditions standards sont : une pression de 1 atm, une température de 25 °C, soit 298 K, une concentration des solutés de = 1 mol/l, un pH de 7, soit une concentration en ions = 10−7 mol/l, une concentration de l'eau de 55,5 mol/l, qui est considérée comme constante et n'apparait pas dans les constantes d'équilibre. L'enthalpie libre standard de réaction d'un système biologique est notée . Les activités des constituants sont écrites en fonction de leur concentration molaire (en mol/l) : (sans dimension). Puisque = 1 mol/l, par simplification on écrit . Ainsi l'enthalpie libre de réaction, notée , s'écrit-elle : . Voir Emmanuel Jaspard, université d'Angers - UFR Sciences, « Biochimie structurale et métabolique », sur biochimej.univ-angers.fr (consulté le ).


























![{\displaystyle \left[{\rm {H}}^{+}\right]}](https://img.franco.wiki/i/f82441a4b367d0aba4c7ba17429a8c16246a457c.svg)
![{\displaystyle \left[{\rm {C}}_{k}\right]}](https://img.franco.wiki/i/faa352819455f2086ca3d7f2dbd9e6e38b0c5ced.svg)
![{\displaystyle a_{k}=\left[{\rm {C}}_{k}\right]/c^{\circ }}](https://img.franco.wiki/i/c619d5714f4979e55538f9a037dfd6b625fbc07a.svg)
![{\displaystyle a_{k}=\left[{\rm {C}}_{k}\right]}](https://img.franco.wiki/i/5c07452f01860dd1ab2a5fb3bbcfff18e0a08882.svg)

![{\displaystyle \Delta _{\text{r}}G^{\prime }=\Delta _{\text{r}}G^{\circ \prime }+RT\,\ln \!\left(\prod _{k=1}^{N}{\left[{\rm {C}}_{k}\right]}^{\nu _{k}}\right)}](https://img.franco.wiki/i/b8641956f0f1b24b174e0f4f6c2cba9839ab64de.svg)
Références
- ↑ SABIX - Société des amis de la bibliothèque et de l'histoire de l'École polytechnique - Bulletin no 23 (avril 2000) - Biographie de Berthollet.
- ↑ SABIX - Société des amis de la bibliothèque et de l'histoire de l'École polytechnique - Bulletin no 24 (août 2000) - Berthollet et la théorie des affinités.
- 1 2 3 4 Kostas Ganaras et Alain Dumon, « La construction du concept d’équilibre chimique », L'Actualité chimique, , p. 38-47 (lire en ligne [PDF], consulté le ).
- 1 2 3 Jacques Schwartzentruber, École nationale supérieure des mines d'Albi-Carmaux, « Effet de la composition globale du système sur le déplacement de la réaction », sur nte.mines-albi.fr, (consulté le ).
- 1 2 3 4 Corriou 1985, p. 20.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 14 (en) J. De Heer, « The Principle of Le Châtelier and Braun », Journal of Chemical Education, vol. 34, no 8, , p. 375-380 (DOI 10.1021/ed034p375).
- ↑ Encyclopædia universalis - affinité chimique.
- ↑ Jean Hertz, « Historique en grandes enjambées de la thermodynamique de l’équilibre », J. Phys. IV France, vol. 122, , p. 3-20 (DOI 10.1051/jp4:2004122001, lire en ligne [PDF], consulté le ).
- ↑ (en) Gilbert Newton Lewis et Merle Randall, Thermodynamics and the free energy of chemical substances, McGraw-Hill Book Company Inc., .
- ↑ Ilya Prigogine et Raymond Defay, Traité de thermodynamique, conformément aux méthodes de Gibbs et de Donder, Desoer, .
- 1 2 3 Gilles Lefebvre, Notions de chimie du pétrole, Paris, Éditions Technip, coll. « École nationale supérieure du pétrole et des moteurs », , 123 p. (ISBN 2-7108-0512-X, lire en ligne), p. 96.
- 1 2 3 4 5 6 7 8 Inaki de Aguirre et Marie-Anne Van de Wiel, Introduction à la chimie générale : Chimie minérale, t. 2, De Boeck Supérieur, , 518 p. (ISBN 2-8041-0720-5, lire en ligne), p. 26-29.
- 1 2 3 Paul Depovere, Chimie générale, Bruxelles, De Boeck Université, , 112 p. (ISBN 2-8041-5029-1, lire en ligne), p. 50-52.
- 1 2 3 4 Soustelle 2015, p. 32.
- 1 2 3 Trambouze et al. 2002, p. 1-2.
- 1 2 3 4 Soustelle 2015, p. 17.
- 1 2 Trambouze et al. 2002, p. 11.
- 1 2 Soustelle 2015, p. 21-22.
- 1 2 3 4 5 6 Mesplède 2004, p. 45.
- 1 2 Jacques Schwartzentruber, École nationale supérieure des mines d'Albi-Carmaux, « Caractérisation de l'équilibre chimique », sur nte.mines-albi.fr, (consulté le ).
- ↑ Alain Demolliens et Corinne Gauthier, Chimie générale : Tout le Cours - PC - PC*, Nathan, coll. « classe prépa », 275 p. (ISBN 978-2-09-812261-1, lire en ligne), p. 35.
- ↑ Mesplède 2004, p. 82.
- ↑ Bruno Fosset, Jean-Bernard Baudin et Frédéric Lahitète, Chimie tout-en-un PC-PC*, Dunod, , 3e éd., 1008 p. (ISBN 978-2-10-076866-0, lire en ligne), p. 124-125.
- 1 2 Odile Durupthy, Alain Jaubert, André Durupthy et Jacques Estienne, Chimie 2e année MP-MP*/PT-PT* : Cours avec exercices corrigés, Hachette Éducation, coll. « H Prépa », , 256 p. (ISBN 978-2-01-181760-0, lire en ligne), p. 87-89.
- 1 2 3 Soustelle 2015, p. 22.
- ↑ Mesplède 2004, p. 46.
- 1 2 3 4 Mesplède 2004, p. 47.
- ↑ Jacques Schwartzentruber, École nationale supérieure des mines d'Albi-Carmaux, « Énoncé de la loi d'action de masse », sur nte.mines-albi.fr, (consulté le ).
- ↑ Université des sciences en ligne (Unisciel), « Calcul de l'enthalpie libre standard de réaction », sur uel.unisciel.fr (consulté le )
- ↑ Trambouze et al. 2002, p. 8.
- ↑ Claude Friedli, Chimie générale pour ingénieur, PPUR presses polytechniques, coll. « Cahiers de chimie », , 747 p. (ISBN 9782880744281, lire en ligne), p. 191.
- ↑ Anthony Bourgeais, chap.4 - Équilibres chimiques, Nathan (ISBN 9782098122222, lire en ligne), p. 100.
- ↑ Jean-Noël Foussard, Edmond Julien, Stéphane Mathé et Hubert Debellefontaine, Thermodynamique : Applications aux systèmes physicochimiques. Cours et exercices corrigés, Dunod, , 278 p. (ISBN 9782100728947, lire en ligne), p. 232.
- ↑ Richard Mauduit et Éric Wenner, Chimie générale en 30 fiches, Dunod, coll. « Express BTS », , 160 p. (ISBN 9782100539482, lire en ligne), p. 36.
- ↑ Henri Fauduet, Principes fondamentaux du génie des procédés et de la technologie chimique, Lavoisier, , 2e éd., 800 p. (ISBN 9782743064556, lire en ligne), p. 191.
- 1 2 Mesplède 2004, p. 41.
- ↑ Infelta et al. 2006, p. 202-203 ; 375-377.
- ↑ Mesplède 2004, p. 41-42.
- ↑ Bruno Fosset, Jean-Bernard Baudin et Frédéric Lahitète, Chimie : Exercices et annales PC-PC*, Dunod, , 152 p. (ISBN 9782100753383, lire en ligne), p. 105-106;118.
- 1 2 Soustelle 2015, p. 61-62.
- 1 2 3 4 5 6 7 Corriou 1985, p. 19.
- ↑ Université du Québec à Chicoutimi (UQAC), « Cours de cinétique chimique : Chapitre I, Caractères généraux de la cinétique des réactions homogènes, Paragraphe 6, Influence de la température sur la vitesse de réaction », sur uqac.ca (consulté le ).
- ↑ Université du Québec à Chicoutimi (UQAC), « Cours de cinétique chimique : Chapitre II, Théorie des vitesses de réaction, Paragraphe 4, L'entropie de réaction », sur uqac.ca (consulté le ).
- ↑ Michel Soustelle, Cinétique chimique : éléments fondamentaux, Paris, Hermès Science Publications-Lavoisier, , 235 p. (ISBN 978-2-7462-3002-6, lire en ligne), p. 68.
- 1 2 3 Mesplède 2004, p. 53-54.
- ↑ Article de Henry Le Chatelier commenté sur Bibnum : Henry Le Chatelier, « Sur un énoncé général des lois des équilibres chimiques », Comptes-rendus de l’Académie des sciences, vol. 99, , p. 786-789 (lire en ligne).
- ↑ Jacques Maddaluno, Véronique Bellosta, Isabelle Chataigner, François Couty, Anne Harrison-Marchand, Marie-Claire Lasne, Chrystel Lopin-Bon et Jacques Rouden, Chimie organique : Exercices et méthodes : Fiches de cours et 500 QCM et exercices d'entrainement corrigés, Dunod, , 354 p. (ISBN 978-2-10-076594-2, lire en ligne), p. 90.
- ↑ Corriou 1985, p. 18.
- 1 2 3 4 5 6 7 8 Joan Josep Solaz-Portolés, Université de Valence, « Pourquoi continuer à apprendre le principe de Le Chatelier ? », Bulletin de l'union des physiciens, vol. 755, no 87, , p. 895-908 (lire en ligne [PDF], consulté le ).
- ↑ Soustelle 2015, p. 37.
- 1 2 3 4 Fabien Caillez, Université Paris-Sud, « Cours de thermodynamique, électrochimie et thermodynamique hors équilibre » [PDF], sur hebergement.u-psud.fr (consulté le ), p. 22-23.
- ↑ Mesplède 2004, p. 87.
- ↑ Alfred-Marie Liénard, « Thermodynamique. Quelques particularités des déplacements de l’équilibre et stabilité », Annales de la faculté des sciences de Toulouse, 4e série, t. 8, , p. 1-58 (lire en ligne, consulté le ).
- ↑ (en) Herbert B. Callen, Thermodynamics and an Introduction to Thermostatistics, New York, John Wiley & Sons, , 2e éd., 512 p. (ISBN 978-0-471-86256-7 et 978-0-471-61056-4, OCLC 11916089, lire en ligne), p. 210-214.
- ↑ Liénard 1944, p. 45.
- 1 2 Mesplède 2004, p. 89.
- 1 2 3 Soustelle 2015, p. 38.
- 1 2 Kévin Moris, Philippe Hermann et Yves Le Gal, Chimie PCSI, Dunod, coll. « Le compagnon », , 496 p. (ISBN 9782100568260, lire en ligne), p. 429.
- 1 2 3 4 « athermique », dictionnaire Larousse.
- 1 2 3 4 « exothermique », dictionnaire Larousse.
- 1 2 3 4 « endothermique », dictionnaire Larousse.
- 1 2 Geneviève M.L. Dumas et Roger I. Ben-Aïm, Thermodynamique chimique : les fondements, Bréal, coll. « L'indispensable », (ISBN 9782749522449, lire en ligne), « Fiche 18 - Chaleur de réaction », p. 68-71.
- ↑ Mesplède 2004, p. 88.
- ↑ Roger Lamartine et Jean-Pierre Scharff, Estérification, vol. J 5 800, Techniques de l'ingénieur, (lire en ligne), paragraphe 2.2 p. 5.
- 1 2 Mesplède 2004, p. 85.
- ↑ Infelta et al. 2006, p. 205 ; 389-390.
- 1 2 3 4 5 6 Mesplède 2004, p. 90.
- 1 2 3 Infelta et al. 2006, p. 189-190.
- 1 2 3 4 5 6 Soustelle 2015, p. 39-43.
- ↑ Jean-Claude Legrand, Thermodynamique chimique : les applications, Bréal, coll. « L'indispensable », (ISBN 978-2-7495-2241-8, lire en ligne), p. 88.
- ↑ Académie de Grenoble, « Le CO2 », sur ac-grenoble.fr (consulté le ).
- 1 2 3 4 5 6 Mesplède 2004, p. 91.
- ↑ Grande Encyclopédie Larousse, « Équilibre chimique », sur Larousse.fr, 1971-1976 (consulté le ), p. 4925.
- 1 2 3 4 5 Mesplède 2004, p. 92.
- 1 2 3 Soustelle 2015, p. 43-44.
- ↑ Élisabeth Bardez, Chimie générale : Chimie des solutions, Dunod, coll. « Mini Manuel », , 3e éd., 272 p. (ISBN 9782100822706, lire en ligne), p. 17.
- 1 2 3 Frédéric Ravomanana, PACES UE1 Chimie générale, Ediscience, , 5e éd., 224 p. (ISBN 978-2-10-078317-5, lire en ligne), p. 82.
- 1 2 Richard Mauduit et Éric Wenner, Chimie générale en 30 fiches, Dunod, , 160 p. (ISBN 978-2-10-053948-2, lire en ligne), p. 156.
- 1 2 3 4 Jean-Léon Houzelot, Réacteurs chimiques polyphasés, vol. J 4 012, Techniques de l'Ingénieur (lire en ligne), p. 6.
- ↑ Trambouze et al. 2002, p. 12.
- ↑ Soustelle 2015, p. 44-45.
- 1 2 3 Infelta et al. 2006, p. 172.
- 1 2 Peter H Raven, Georges B Johnson, Kenneth A Mason, Jonathan B Losos et Susan R Singer (trad. de l'anglais par Jules Bouharmont, Pierre L Masson, Charles Van Hove), Biologie [« Biology »], De Boeck Supérieur, , 4e éd., 1400 p. (ISBN 9782807330207, lire en ligne), II. Biologie cellulaire, chap. 6 (« Énergie et métabolisme »), p. 107-121.
- ↑ Donald Voet et Judith G. Voet (trad. de l'anglais par Lionel Domenjoud), Biochimie [« Biochemistry »], De Boeck Supérieur, , 3e éd., 1784 p. (ISBN 9782804171018, lire en ligne), p. 579.
- ↑ Kathleen M Botham, Anthony Weil, Victor W Rodwell, Peter J Kennelly et David A Bender (trad. de l'anglais par Lionel Domenjoud), Biochimie de Harper [« Harper's Illustrated Biochemistry »], De Boeck Supérieur, , 6e éd., 840 p. (ISBN 9782807307247, lire en ligne), p. 114.
Bibliographie
- J. Willard Gibbs (trad. Henry Le Chatelier, accessible sur Gallica), Équilibre des systèmes chimiques, Paris, G. Carré et C. Naud, (lire en ligne).
- Jean-Pierre Corriou, Thermodynamique chimique : Équilibres thermodynamiques, vol. J 1028, Techniques de l'ingénieur, coll. « base documentaire Thermodynamique et cinétique chimique, pack Opérations unitaires. Génie de la réaction chimique, univers Procédés chimie - bio - agro », (lire en ligne), p. 1-31.
- P. Infelta et M. Graetzel, Thermodynamique : Principes et Applications, BrownWalker Press, , 484 p. (ISBN 978-1-58112-995-3, lire en ligne), chapitres 9 et 10.
- J. Mesplède, Chimie PSI : Cours - Méthodes - Exercices résolus, Bréal, coll. « Les nouveaux précis Bréal », , 350 p. (ISBN 978-2-7495-2063-6, lire en ligne), partie I - Thermodynamique ; Chapitre 2 - Équilibres chimiques pp. 37-80 ; Chapitre 3 - Déplacement des équilibres pp. 81-116.
- Michel Soustelle, Équilibres chimiques, vol. 4, Londres, ISTE Group, coll. « Génie des procédés / Thermodynamique chimique approfondie », , 196 p. (ISBN 978-1-78405-103-7, lire en ligne).
- Pierre Trambouze et Jean-Paul Euzen, Les réacteurs chimiques : De la conception à la mise en œuvre, Éditions OPHRYS, coll. « Publications de l'Institut français du pétrole », (ISBN 2-7108-1121-9, lire en ligne).
Liens externes
- « Les équilibres chimiques », sur sciences-en-ligne.com (consulté le ).
- Jacques Schwartzentruber, École nationale supérieure des mines d'Albi-Carmaux, « Les équilibres chimiques », sur nte.mines-albi.fr, (consulté le ).
Voir aussi
Exemples d'équilibres
- Aldolisation
- Autoprotolyse
- Estérification / Hydrolyse
- Équilibre calco-carbonique
- Équilibre de Boudouard
- Équilibre de Schlenk
- Précipitation chimique / Solubilité
- Réaction acido-basique
- Réaction d'hydratation / Réaction de déshydratation
- Réaction d'oxydoréduction
- Réaction du gaz à l'eau
- Sulfonation
Articles connexes
- Activité chimique
- Affinité chimique
- Constante d'équilibre
- Détermination des constantes d'équilibre
- Deuxième principe de la thermodynamique
- Enthalpie libre
- Équilibre thermodynamique
- Fugacité
- Grandeur de réaction
- Loi d'action de masse
- Potentiel chimique
- Relations de Kirchhoff