| Encéphalopathie Spongiforme Bovine (ESB) | |
| Chromosome | Chromosome 20 humain 20p13 |
|---|---|
| Gène | PRNP |
| Mutation | dans la protéine Prion |
| Liste des maladies génétiques à gène identifié | |
L’encéphalopathie spongiforme bovine (ESB), également appelée « maladie de la vache folle » (Bovine spongiform encephalopathy [BSE] en anglais), est une infection dégénérative du système nerveux central des bovins. C'est une maladie mortelle, analogue à la tremblante des ovins et des caprins, causée par un agent infectieux moléculaire d'un type particulier (ni virus, ni bactérie), appelé protéine prion.
Une épizootie d'ESB a touché le Royaume-Uni, et dans une moindre mesure quelques autres pays, entre 1986 et les années 2000, infectant plus de 190 000 animaux, sans compter ceux qui n'auraient pas été diagnostiqués. Cette crise de la vache folle trouve son origine dans l'utilisation pour l'alimentation des bovins de farines animales, obtenues à partir de parties non consommées des carcasses bovines et de cadavres d'animaux. L'épidémie a pris une tournure particulière quand les scientifiques se sont aperçus début 1996 de la possibilité de transmission de la maladie à l'Homme par le biais de la consommation de produits carnés. La maladie a fait 231 victimes humaines jusqu'en 2016, touchées par des symptômes proches de la maladie de Creutzfeldt-Jakob, une maladie de même nature que l'ESB.
Dès la déclaration officielle par le gouvernement du Royaume-Uni, le 21 mars 1996, de la transmission de la maladie bovine à l'Homme, les médias se sont rapidement emparés de l'affaire et l'ont relayée devant le grand public, engendrant une crise sans précédent. Cette crise fut à la fois éthique, avec la prise de conscience des consommateurs de certaines pratiques courantes en élevage jusqu'alors ignorées de ceux-ci, telles que l'utilisation des farines animales pour l'alimentation, et économique du fait de la chute de consommation de viande bovine qui en suivit et du coût des différentes mesures adoptées.
Il n'existe actuellement aucun traitement curatif contre la maladie, et elle n'a pu être enrayée que par des mesures prophylactiques.
Épidémiologie
La maladie a été identifiée pour la première fois en Grande-Bretagne en 1986.
Symptômes
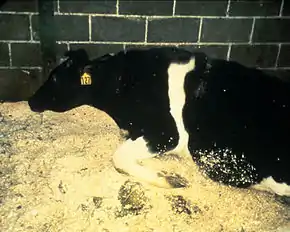
L’ESB affecte le cerveau et la moelle épinière des bovins. Elle provoque des lésions cérébrales qui se caractérisent par des altérations à allure spongieuse visibles au microscope optique, correspondant à des neurones qui se sont vacuolisés. Il y a une perte de neurones plus ou moins importante, et une multiplication des astrocytes, des cellules du cerveau à fonction immunitaire. Les agents pathogènes s'amassent pour former des plaques amyloïdes caractéristiques, quoique moins présentes que pour d'autres encéphalopathies spongiformes transmissibles[1]. Les symptômes extérieurs apparaissent généralement 4 à 5 ans après la contamination, et toujours sur des animaux de plus de 2 ans (entre 3 et 7 ans généralement). Ils se manifestent au début par une modification du comportement de l'animal, qui peut parfois donner des coups de pied, manifester une appréhension et une hypersensibilité aux stimulations externes (bruit, toucher, éblouissement) et s'isoler du reste du troupeau. L'animal atteint voit généralement sa production laitière et son poids décroître, alors que son appétit ne diminue pas. L'évolution peut durer d'une semaine à un an, les différentes phases de la maladie étant d'une durée variable d'un animal à un autre. Au stade ultime d'évolution, l'animal a de véritables troubles de la locomotion. Il perd fréquemment l'équilibre, sans parvenir parfois à se relever. Du point de vue physiologique, on observe une tachycardie et une absence de fièvre. Cependant, l'apparition de ces symptômes ne permet pas à coup sûr de détecter un cas d'ESB. En effet, les troubles locomoteurs, comme la tétanie d'herbage, sont fréquents chez les bovins, et le diagnostic de la maladie est donc difficile[2].
Agent pathogène non conventionnel
La nature réelle de l'agent infectieux fait débat. La théorie maintenant largement admise par la communauté scientifique est celle du prion, une protéine qui, dans le cas de la maladie, adopte une conformation anormale pouvant se transmettre à d'autres protéines prions saines. Une théorie alternative est l'agent viral, qui expliquerait plus facilement la capacité de l’agent à générer de multiples souches.[3] Le prion (PRoteinasceous Infectious ONly) est une sialoglycoprotéine de 253 acides aminés. Les sialoglycoprotéines interviennent dans l’adhésion cellulaire. C'est une forme dite résistante de cette protéine qui est responsable de la maladie. Cette forme diffère de la forme normale uniquement par sa conformation, qui la rend particulièrement hydrophobe, ce qui explique la formation des agrégats résistants aux protéases et l’accumulation de la protéine infectieuse dans la cellule. La protéine modifiée pénètre dans la cellule par endocytose ou par le biais du récepteur spécifique du prion normal, la protéine LRP (Laminin Receptor Protein), qui semble intervenir dans le processus puisque sa concentration augmente avec la contamination. Une fois la protéine anormale entrée dans la cellule, elle transforme les prions normaux en prions résistants. Les protéases n'étant plus capables de les détruire, ces protéines s'accumulent pour finir par provoquer la mort du neurone, et ainsi la formation de plaques amyloïdes[4]. Comme le prion est une protéine, il n'a pas de métabolisme propre et il est donc résistant à la congélation, à la dessiccation et à la chaleur aux températures normales de cuisson, même celles atteintes pour la pasteurisation et la stérilisation[5]. En effet, pour être détruit, le prion doit être chauffé à une température de 133 °C pendant 20 minutes à 3 bars de pression[6].
Origines de l'épidémie

On ne sait pas réellement comment est apparu l'agent pathogène de l'ESB. Deux hypothèses dominent.
La première est une contamination interspécifique à partir d'une maladie proche, la tremblante du mouton. Cette possibilité a été expérimentalement prouvée mais les troubles cliniques et neuropathologiques diffèrent dans les deux cas. Ce constat a conduit à une seconde hypothèse qui est que la maladie serait endémique à l'espèce bovine et très faiblement répandue avant qu'elle ne soit amplifiée au milieu des années 1980[5]. La description dans une revue vétérinaire de 1883 d'un cas de tremblante chez un bovin est un argument utilisé par les défenseurs de cette théorie, bien que ce cas puisse correspondre à une toute autre maladie neurologique[7].
D'autres théories plus ou moins crédibles ou discutées sont régulièrement débattues. Ainsi :
- le professeur Roger Morris de l'université Massey à Palmerston North (Nouvelle-Zélande) estime que des antilopes contaminées par une maladie proche dans des parcs anglais, puis réduites en farines pour l'alimentation du bétail après leur mort pourraient être une origine de l'ESB[8] ;
- pour le scientifique John Williams, une pollution au bromure de méthyle (produit mutagène) en 1963 dans le Kent aurait fait muter le prion d'une vache en une forme anormale ;
- l'anglais Mark Purdey a au début des années 2000 posé l'hypothèse que des infrasons[9] et des microcristaux anormaux[10],[11],[12] aient pu induire une mutation de la protéine prion en une forme déviante, capable de se transmettre et se « dupliquer » ; il a ensuite aussi proposé une explication pour la CWD (maladie proche qui se développe chez les cervidés en Amérique du Nord) ou d'autres formes d'encéphalopathies spongiformes transmissibles[13].
On ne saura peut-être jamais avec certitude comment et quand est exactement apparu l'agent pathogène responsable de la maladie[7], mais concernant sa circulation, le recyclage des carcasses d'animaux par les équarrisseurs semble en cause. Les parties d'os et de viande non utilisées dans l'alimentation humaine ; les animaux morts ramassés en ferme par les services d'équarrissage constituent les principaux déchets de l'industrie de la viande bovine, ils sont séparés de leurs graisses par cuisson avant d'être réduits en farine. Avant l'apparition de l'ESB, les farines animales[note 1] ont été très utilisées dans l'alimentation du bétail et d'autres animaux, car énergétiques et riches en minéraux et protéines, et bien digérées par les ruminants. Elles étaient donc très utilisées chez les bovins, notamment chez les vaches laitières[14]. C'est la consommation par les bovins de farines animales issues de tissus calcinés provenant de bovins ou d'ovins (suivant l'hypothèse retenue), comme la cervelle et la moelle épinière, et contaminés par l'agent de l’ESB qui est probablement responsable de l'apparition de l'épidémie[5].
Initialement, ces farines étaient stérilisées à hautes températures et une étape d'extraction des graisses par solvants organiques permettaient, sans que personne le soupçonne, de détruire d'éventuels prions pathogènes. Mais en 1981, les températures de stérilisation ont été abaissées et l'étape d'extraction des graisses par solvants a été éliminée. Cette simplification du protocole visait à améliorer la rentabilité de la filière, d'une part en préservant mieux les protéines contenues dans les farines, d'autre part en diminuant les achats de solvants et d'énergie dont les coûts avaient beaucoup augmenté après les deux chocs pétroliers de 1973 et 1979. De plus un accident de manipulation de solvant dans une des principales usines anglaises de fabrication de farines animales avait entraîné un renforcement des mesures de sécurité dont le coût était élevé, ce qui a aussi encouragé cette modification des pratiques qui semble avoir causé l'épidémie. Le prion a alors pu être distribué dans les farines animales à grande échelle via les aliments du bétail, et les animaux contaminés et abattus étaient à leur tour réduits en farines, ce qui aggravait le phénomène[4].
Une voie de contamination mère-veau est aussi soupçonnée. Elle pourrait représenter jusqu'à 10 % des contaminations[2].
Cas NAIF et rémanence de l'épidémie d'ESB
Rien qu'en France, 847 cas NAIFs (Nés Après l'Interdiction des Farines) avaient été recensés au 1er juin 2006, avec une répartition géographiquement et temporellement hétérogène[15],[16]. 85 % des cas recensés de 1997 à 2006 (animaux nés pour la plupart entre 1993 et 1995) ont dû être classés comme « NAIF », mais sans explication claire du mécanisme exact de transfert des prions pathogènes.
Pour expliquer ces cas et la poursuite à bas bruit de la zoonose en dépit de mesures draconiennes (bien après l'interdiction des farines animales en France en 1990 et de nombreux contrôles), une éventuelle troisième voie de contamination est recherchée, mais pas encore trouvée. Parmi les rares hypothèses crédibles on recense :
- une contamination épisodique par la nourriture artificielle donnée aux bovins : plusieurs études[17],[18],[19],[20] ont montré que le risque de cas NAIF d'ESB augmentait significativement chez les bovins ayant consommé des aliments industriels durant les deux premières années de vie, et plus encore durant leurs premiers mois de vie. De plus, l'achat de lactoremplaceurs pour bovins ou — plus curieusement — l’achat d'aliments du commerce pour volailles (dans une exploitation faisant également de l'élevage bovin) augmentait aussi significativement le risque d'apparition d'un cas NAIF d'ESB, ce qui invite à envisager qu'il y ait eu des contaminations croisées[21] ;
- une contamination médiée par des acariens du fourrage : ce phénomène a été observé une fois pour la tremblante du mouton. Cette hypothèse, et celles basées sur un agent de transmission extérieur, a été jugée peu vraisemblable car seul le système nerveux central semble contaminant chez les bovins et le prion n’est pas réputé excrété par les vaches malades ;
- un facteur (ou cofacteur) bactérien a été évoqué en 2004, dont par L Broxmeyer[22]. La bactérie en cause pouvant être un spiroplasme pour F.O Bastian (2005)[23] ou Brucella abortus pour Watarai (2004)[24], à moins que le prion ne soit dans ces cas un simple récepteur de ces pathogènes. Une protéine chaperon (dite protéine X, non identifiée chez les mammifères) pourrait jouer un rôle dans la conversion de la PrPc en PrPSc selon Telling et al. (1995)[25]. La région C-terminale de la PrP serait le site potentiel de liaison avec cette protéine X[25],[26],[27]. Des protéines chaperons (ex Hsp104 et GroEL respectivement chez les levures et les bactéries) stimulent effectivement in vitro la formation de protéines prion « scrapie » dans un système de conversion in vitro acellulaire[28] mais la protéine X si elle existe n'a pas encore été identifiée chez l'Homme ;
- une contamination par de l’eau polluée par des centres d’équarrissage ou via le sol où ont été épandues des matières fertilisantes à base de farines animales, sans preuve tangible selon l'AFSSA[29].
Transmission
Chez les humains
Il existe une forme d'encéphalopathie subaiguë spongiforme transmissible (ESST) spécifique à l'Homme, connue sous le nom de maladie de Creutzfeldt-Jakob (MCJ) qui est une dégénérescence du système nerveux central caractérisée par l'accumulation d'un prion. La période d'incubation se compte en années, voire en décennies avant qu'apparaissent des troubles de l'équilibre et de la sensibilité, puis une démence. L'issue est systématiquement fatale à échéance d'approximativement un an. Cette maladie a plusieurs causes : la plupart des cas sont dits sporadiques, car l'origine est inconnue. Il existe également une transmission héréditaire (10 % des cas) et des contaminations iatrogéniques (c'est-à-dire dues à une intervention médicale) liées à l'utilisation d'hormone (comme dans l'affaire de l'hormone de croissance en France) ou de greffes de tissus cérébraux (dure-mère) issus de cadavres de malades, ou encore par l'utilisation d'instruments de chirurgie mal décontaminés (électrodes).
Les décès par la maladie de Creutzfeldt-Jakob d'éleveurs entre 1993 et 1995 avaient les premiers inquiété les scientifiques sur la probabilité de la transmission de l'ESB à l'Homme, mais ils avaient alors conclu à des cas sporadiques sans lien avec la maladie animale[30]. C'est en 1996, lorsque deux Britanniques habitant au nord de Londres moururent d'une maladie qui semblait être à première vue la maladie de Creutzfeldt-Jakob que tout commença réellement. Stephen Churchill et Nina Sinnott, qui avaient respectivement 19 et 25 ans, étaient anormalement jeunes pour contracter cette maladie qui touche exclusivement les personnes âgées. C'est cela qui mit les chercheurs sur la voie d'une nouvelle maladie, notée nvMCJ, pour « nouveau variant de la maladie de Creutzfeldt-Jakob ». Rapidement, un lien est soupçonné entre l'ESB, maladie animale et la nouvelle variante de la maladie de Creutzfeldt-Jakob, maladie humaine[note 2]. Ce lien a été démontré en laboratoire en comparant les plaques amyloïdes présentes dans le cerveau de singes auxquels on avait inoculé la maladie et celles des jeunes gens morts de la maladie, qui se sont révélées strictement identiques. La forme humaine de l'ESB ressemble sous ses grands traits à la maladie de Creutzfeldt-Jakob, mais s'en distingue par quelques différences cliniques et anatomiques. Ainsi, elle affecte des patients plus jeunes (âge moyen de 29 ans, contre 65 ans pour la maladie classique) et a une évolution relativement plus longue (médiane de 14 mois au lieu de 4,5 mois)[5]. Les premiers symptômes sont des troubles neuropsychiatriques, parfois suivis d'une dépression brutale ou d'une profonde anxiété. Le malade est fatigué, a des douleurs parfois importantes. Au bout de quelque temps dans l'évolution de la maladie, qui n'est pas continue dans le temps et peut connaître des périodes de stabilité relative, les troubles neurologiques se font plus pressants. Le malade perd la mémoire et le sens de l’orientation et son comportement devient anormal. Puis, il ne parvient plus à coordonner ses mouvements qui sont brusques et parfois involontaires. Il finit par décéder de la maladie. La maladie peut être transmise à l'Homme s'il consomme de la viande ou des tissus issus d'animaux contaminés. Après l'ingestion du prion, celui-ci pénètre dans les formations lymphoïdes des intestins, notamment les plaques de Peyer, dans lesquelles il peut se répliquer. Il progresse ensuite dans les nerfs au rythme d’un millimètre par jour, pour finalement atteindre la moelle épinière puis le cerveau où il provoque des lésions caractéristiques. Toutefois, certains paramètres de l'infection restent mal connus, comme la dose infectieuse, la durée d'incubation chez l'Homme et la manière avec laquelle le prion pénètre dans les nerfs[7].
En 2016, on estime que la maladie a fait 231 victimes, dont 178 au Royaume-Uni, 27 en France, 5 en Espagne, 4 en Irlande et aux États-Unis, 3 aux Pays-Bas et en Italie, 2 au Portugal et au Canada, et 1 au Japon, à Taïwan et en Arabie saoudite[31].
Aux autres animaux domestiques

Lors de l'apparition de la maladie, on lui a souvent donné une origine ovine du fait de sa ressemblance avec la tremblante du mouton. Aujourd'hui, cette hypothèse a perdu un peu de son crédit. Toutefois, si on ne connaît pas l'origine exacte de la maladie, il reste certain qu'elle a une forte propension à traverser la barrière de l'espèce. Dès mai 1990, l'épidémie s'étend aux félidés avec la mort d'un chat domestique victime de la maladie, probablement contaminé par la nourriture, les aliments pour chats étant très souvent fabriqués à partir d'abats de bovins[note 3]. L’ESB a contaminé des antilopes et d’autres ruminants sauvages dans les zoos britanniques, ainsi qu’un certain nombre de carnivores : lions, tigres, pumas, guépards. Elle est également facilement transmissible à d'autres espèces de bovidés sauvages comme le bison. On a pu expérimentalement constater la transmission possible à d'autres mammifères, dont la souris, le porc et certains singes, en recourant toutefois à des méthodes très invasives (injection dans le cerveau).
Il existe différentes ESST qui touchent d'autres ruminants, sans qu'aucun lien direct ne soit prouvé avec l'ESB. Parmi ces maladies, on compte notamment la tremblante du mouton, qui touche les petits ruminants. Aucun cas de transmission de cette maladie à l'Homme n'a jamais été répertorié. La cachexie chronique/Chronic Wasting Disease (CWD) fait également partie de la famille des ESST. Elle affecte principalement les États-Unis et dans une moindre mesure le Canada. Cette maladie à prion décime, que ce soit en fermes d'élevage ou dans la nature, les cervidés. Ceux-ci présentent les mêmes symptômes que les bovidés atteints de la maladie de la vache folle, mais la maladie évolue plus rapidement vers la mort[32]. Le ministère de l'agriculture des États-Unis (USDA) a organisé de gigantesques battues, requérant l'aide de chasseurs, dans le but d'éradiquer ce mal 10 000 cervidés abattus et incinérés au Colorado en 2004), qui se révèle incontrôlable. Plusieurs chasseurs ont été atteints de la maladie de Creutzfeldt-Jakob, mais le gouvernement américain se refuse à faire un lien avec la « maladie du cerf fou », tout comme avec les récents cas de maladie de la vache folle découverts aux États-Unis (deux) en 2004-05 ou au Canada.
L'Agence française de sécurité sanitaire des aliments (AFSSA) a publié en mars 2005 un avis confirmant définitivement le risque d'ESB chez les petits ruminants (chèvres et moutons). Chez ces deux espèces, le risque de transmission à l'Homme peut être plus élevé, car, outre la viande, le lait peut être contaminé. L'Afssa juge insuffisantes les mesures de précaution prises ; le lait des troupeaux suspects n'étant pas testé, et une partie seulement des cadavres d'animaux suspects faisant l'objet de recherches sur les prions[33].
Par contre, un certain nombre d'espèces semblent insensibles à la maladie. C'est notamment le cas des oiseaux, qui ne souffrent d'ailleurs d'aucune ESST, probablement du fait de leur courte durée de vie. Le chien, le lapin, le cheval et le poisson ne sont pas non plus concernés par ce type de maladies neurologiques[7].
Conséquences et crise
Relayées par les médias, les informations sur les risques pour la santé humaine véhiculés par la maladie provoquent une chute rapide de la consommation de viande, responsable de grandes difficultés économiques dans l'ensemble de la filière viande bovine. Outre la baisse de la consommation, les acteurs de cette filière se voient répercuter les frais liés à l'éradication de la maladie. Ainsi, les éleveurs ont subi de lourdes pertes avec l'abattage systématique de leurs troupeaux, et les abattoirs et transformateurs secondaires ont dû s'adapter à une nouvelle législation.
Notes et références
Notes
- ↑ ou plus précisément des FVO (farines de viande et d'os)
- ↑ C'est le premier cas de transmission d'une ESST animale à l'Homme. En effet, on n'a jamais démontré que la tremblante du mouton, connue déjà depuis de nombreuses années, pouvait se transmettre à l'Homme.
- ↑ Les consommateurs britanniques n'apprécient pas tellement les abats.
Références
- ↑ F. Lantier et al., « Le diagnostic des encéphalopathies spongiformes chez les ruminants », Productions animales, INRA, vol. Hors-série, , p. 79-86.
- 1 2 Institut de l'Élevage, Maladies des bovins : manuel pratique, France Agricole, , 540 p. (ISBN 978-2-85557-048-8 et 2-85557-048-4).
- ↑ (en) Laura Manuelidis, « A 25 nm Virion Is the Likely Cause of Transmissible Spongiform Encephalopathies », Journal of Cellular Biochemistry, .
- 1 2 [PDF]« L’Encéphalopathie Spongiforme Bovine : E.S.B. », sur univ-brest.fr (consulté le ).
- 1 2 3 4 « Encéphalopathie spongiforme bovine (ESB) », Organisation mondiale de la santé, (consulté le ).
- ↑ Françoise Pradier, « 8 questions que vous pourriez poser à votre boucher », sur doctissimo (consulté le ).
- 1 2 3 4 Gérard DERIOT et Jean BIZET., « Rapport de la commission d’enquête sur les conditions d’utilisation des farines animales dans l’alimentation des animaux d’élevage et les conséquences qui en résultent pour la santé des consommateurs », sur Sénat, (consulté le ).
- ↑ (en) « BSE origins 'linked to antelope' », BBC (consulté le ).
- ↑ Purdey M (2003) Does an infrasonic acoustic shock wave resonance of the manganese 3+ loaded/copper depleted prion protein initiate the pathogenesis of TSE ; Medical hypotheses, 60(6), 797-820.
- ↑ Purdey M (2005) Metal microcrystal pollutants; the heat resistant, transmissible nucleating agents that initiate the pathogenesis of TSEs?. Medical hypotheses, 65(3), 448-477.
- ↑ Purdey M (2004) The environmental origins of TSEs: the ferrimagneto-prion theory. Townsend Letter for Doctors and Patients, (252), 83-89.
- ↑ Purdey, M. (2006). Auburn university research substantiates the hypothesis that metal microcrystal nucleators initiate the pathogenesis of TSEs. Medical hypotheses, 66(1), 197-199.
- ↑ Purdey, M. (2004). Elevated silver, barium and strontium in antlers, vegetation and soils sourced from CWD cluster areas: Do Ag/Ba/Sr piezoelectric crystals represent the transmissible pathogenic agent in TSEs?. Medical hypotheses, 63(2), 211-225.)
- ↑ Alain Sousa, « Le problème des farines animales », sur doctissimo (consulté le ).
- ↑ D. Abrial, D. Calavas, N. Jarrige, C. Ducrot, « Spatial heterogeneity of risk of BSE in France following the ban of meat and bone meal in cattle feed », Preventive Veterinary Medicine, vol. 67, 2004, p. 69-82 (résumé).
- ↑ C. Ducrot, D. Abrial, D. Calavas, & T. Carpenter, « A spatio-temporal analysis of BSE cases born before and after the reinforced feed ban in France », Veterinary research, vol. 36, no 5-6, 2005, p. 839-853.
- ↑ D. Abrial, D. Calavas, N. Jarrige, & C. Ducrot, « Poultry, pig and the risk of BSE following the feed ban in France–a spatial analysis », Veterinary research, vol. 36, no 4, 2005, p. 615-628.
- ↑ N. Jarrige, C. Ducrot, G. Cazeau, E. Morignat, & D. Calavas, « Contamination alimentaire des bovins naïfs atteints d'ESB ; Études complémentaires sur la période de distribution des aliments composés aux bovins et sur le rôle des aliments pour volailles », J. Epidémiol. et santé anim., vol. 49, 2006, p. 45-53.
- ↑ N. Jarrige, C. Ducrot, G. Cazeau, E. Morignat & D. Calavas, « Les aliments pour bovins, principale cause des cas d'ESB nés après l’interdiction des farines animales », dans 13. Rencontres autour des Recherches sur les Ruminants (3R), 2006-12-06/2006-12-07, Paris, FRA. Institut de l’Élevage.
- ↑ N. Jarrige, C. Ducrot, G. Cazeau, E. Morignat, C. La Bonnardière, & D. Calavas, « Case-control study on feed risk factors for BSE cases born after the feed ban in France », Veterinary research, vol. 38, no 3, 2007, p. 505-516.
- ↑ D. Abrial, D. Calavas, N. Jarrige, C. Ducrot, « Poultry pig and the risk of BSE following the feed ban in France - Spatial analysis », Veterinary Research, 2005, vol. 36, p. 615-628.
- ↑ L. Broxmeyer, « Is mad cow disease caused by a bacteria? », Med Hypotheses, vol. 63, no 4, 2004, p. 731-9.
- ↑ F. O. Bastian, « Spiroplasma as a candidate agent for the transmissible spongiform encephalopathies », J Neuropathol Exp Neurol, vol. 64, no 10, 2005, p. 833-8.
- ↑ M. Watarai, « Interaction between Brucella abortus and cellular prion protein in lipid raft microdomains », Microbes Infect, vol. 6, no 1, 2004, p. 93-100.
- 1 2 G. C. Telling, M. Scott, J. Mastrianni, R. Gabizon, M. Torchia, F. E. Cohen, S. J. DeArmond et S. B. Prusiner, « Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein », Cell, vol. 83, no 1, 1995, p. 79-90.
- ↑ K. Kaneko, M. Vey, M. Scott, S. Pilkuhn, F. E. Cohen et S. B. Prusiner, « COOHterminal sequence of the cellular prion protein directs subcellular trafficking and controls conversion into the scrapie isoform », Proc Natl Acad Sci USA, vol. 94, no 6, 1997, p. 2333- 8.
- ↑ M. R. Scott, J. Safar, G. Telling, O. Nguyen, D. Groth, M. Torchia, R. Koehler, P. Tremblay, D. Walther, F. E. Cohen, S. J. DeArmond et S. B. Prusiner, « Identification of a prion protein epitope modulating transmission of bovine spongiform encephalopathy prions to transgenic mice », Proc Natl Acad Sci USA, vol. 94, no 26, 1997, p. 14279-84.
- ↑ K. W. Leffers, J. Schell, K. Jansen, R. Lucassen, T. Kaimann, L. Nagel-Steger, J. Tatzelt & D. Riesner, « The structural transition of the prion protein into its pathogenic conformation is induced by unmasking hydrophobic sites », J Mol Biol, vol. 344, no 3, 2004, p. 839-53.
- ↑ [PDF] « Les risques sanitaires liés aux différents usages des farines et graisses d’origine animale et aux conditions de leur traitement et de leur élimination. », AFSSA, (consulté le ).
- ↑ Séverin Muller, À l'abattoir : travail et relations professionnelles face au risque sanitaire, Quæ, (ISBN 978-2-7592-0051-1 et 2-7592-0051-5).
- ↑ (en) « Variant CJD Cases Worldwide » (consulté le ).
- ↑ (en) E.S. Williams, M.W. Miller, « Chronic wasting disease in deer and elk in North America », Revue scientifique et technique, Office international des épizooties, vol. 21, , p. 305-316.
- ↑ Marianne no 418 du 23 avril 2005.
Annexes
Bibliographie
- Philippe Duneton et Martin Hirsch, L’Affolante histoire de la vache folle, Jacob Duvernet, .
- Jill-Patrice Cassuto, De la maladie de la vache folle à celle de Creutzfeld Jakob, Odile Jacob, .
- Francis Chateauraynaud et Didier Torny, Les Sombres précurseurs. Une sociologie pragmatique de l'alerte et du risque, EHESS, .
- Pierre-Marie Lledo, Histoire de la vache folle, Presses Universitaires de France, .
- Institut de l'Élevage, Maladies des bovins : manuel pratique, France Agricole, , 540 p. (ISBN 978-2-85557-048-8 et 2-85557-048-4).
- [PDF] « L’Encéphalopathie Spongiforme Bovine : E.S.B. », sur univ-brest.fr (consulté le ).
Filmographie
- Documentaire Mort aux Vaches de Frédéric Brunnquell, Arte, 2013.
Articles connexes
- Abattoir
- Prion
- Prion pathogène
- Crise de la vache folle
- Maladie débilitante chronique (encéphalopathie des cervidés), une maladie étrangement proche de celle de la vache folle et découverte dans des élevages du monde entier (Amérique, Europe, Asie).
- Encéphalopathie spongiforme du dromadaire
Liens externes
- Ressources relatives à la santé :
- (en) Diseases Ontology
- (en) Héritage mendélien chez l'humain
- (en) Héritage mendélien chez l'humain
- (en) Medical Subject Headings
- (no + nn + nb) Store medisinske leksikon
- Notices dans des dictionnaires ou encyclopédies généralistes :
- Organisation mondiale de la santé animale, anciennement Office international des épizooties (OIE), site officiel.
- Encéphalopathie spongiforme bovine, sur le site agriculture.gouv.fr.
- Vache folle : l'histoire d'une crise, sur le site de l'Inra.