| Spécialité | Pédiatrie, procédures de chirurgie digestive (en) et gastro-entérologie |
|---|
| CIM-10 | Q43.1 |
|---|---|
| CIM-9 | 751.3 |
| OMIM | 142623 |
| DiseasesDB | 5901 |
| MedlinePlus | 001140 |
| eMedicine | 178493 |
| MeSH | D006627 |
![]() Mise en garde médicale
Mise en garde médicale
La maladie de Hirschsprung est une anomalie de fonctionnement de la partie terminale de l’intestin se traduisant par une constipation ou une occlusion intestinale. Cette anomalie est le résultat de l’absence de développement congénital des cellules neuroganglionnaires assurant la transmission des informations nécessaires à la régulation intestinale.
Cette maladie est considérée comme une neurocristopathie ou maladie dérivant des crêtes neurales.
Cette maladie peut être isolée ou s'inscrire dans un syndrome.
Dans les formes non syndromiques, plusieurs gènes ont été identifiés comme responsables de cette maladie.
Histoire
Maladie décrite par Harald Hirschsprung en 1886. Le mécanisme de la maladie a été découvert par Orvar Swenson qui a également réussi la première intervention en 1948[1].
Autres noms de la maladie
- Aganglionose digestive totale
- Mégacôlon congénital
Incidence
La maladie de Hirschsprung est une maladie génétique rare, touchant une naissance sur 5 000[2], en majorité des garçons (80 %). Il s'agit bien souvent de formes sporadiques (sans antécédent familial).
Description
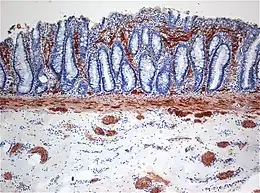
La maladie de Hirschsprung est une malformation congénitale touchant le plexus nerveux intrinsèque qui est anormalement absent de la paroi digestive depuis l’anus jusqu’à une hauteur variable de l’intestin, généralement la partie moyenne du sigmoïde.
Cette maladie est due à l'absence partielle ou totale de ganglions nerveux, dont le rôle est de permettre le bon fonctionnement des muscles de l'intestin, et plus particulièrement du côlon. De l'existence de ce segment aganglionnaire résulte une paralysie intestinale, se traduisant généralement par une occlusion fonctionnelle, parfois simplement par une constipation importante.
La majorité des nouveau-nés atteints ont des retards d’émission du méconium, l'émission ne survenant qu’après plus de 36 heures de vie néonatale. Les signes cliniques sont digestifs : constipation, distension abdominale ou vomissement ou entérocolite. Mais 10 % des diagnostics ne seront faits qu’après l’âge de 1 an (dans les formes non-syndromiques).
Cette maladie a aussi été découverte chez plusieurs espèces animales, dont le rat (ce qui facilite la recherche en laboratoire sur cette maladie). Il semblerait que chez cette espèce l'anomalie génétique comprend également une dépigmentation du pelage, comme chez le cheval.
Formes cliniques
Il existe différentes formes de la maladie de Hirschsprung, dont la gravité dépend de la longueur du segment aganglionnaire. Dans 90 % des cas, il s'agit de la forme « classique » qui touche simplement la terminaison du colon (le rectum et le colon sigmoïde).
Il existe aussi des formes courtes n'intéressant que la partie basse du rectum et à l'opposé des formes longues pouvant atteindre tout le colon voire remonter plus ou moins sur l'intestin grêle.
En fonction de l’étendue de l’atteinte de la maladie au niveau intestinal, on distingue :
- Hirschsprung court segment, localisée au sigmoïde, dans 80 % des cas ;
- Hirschsprung long segment, étendue au côlon, dans 15 % des cas ;
- Hirschsprung totale, localisée à tout l’intestin, dans 5 % des cas.
Diagnostic
La maladie est généralement diagnostiquée dans les toutes premières semaines de vie. À la naissance, l’intestin est bouché. Le nouveau-né a un ballonnement abdominal, souvent considérable, qui peut être accompagné de vomissement bilieux (vert). Les premières selles, qui sont normalement évacuées durant les premières 24 heures, sont retardées anormalement.
À ce stade, on peut diagnostiquer cette maladie grâce au passage en douceur d'une grosse sonde rectale. Celle-ci permet de faire sortir des selles et des gaz en abondance et de déballonner l'enfant. Cela se produit si la sonde a pu monter jusque dans le colon dilaté, soit plus haut que la zone n’ayant pas de cellules neuro-ganglionnaires.
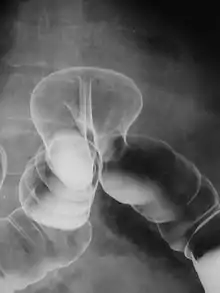
Un lavement opaque radiologique (dit lavement baryté) peut être effectué pour révéler la dilatation du côlon ou de l’intestin grêle au-dessus d’une zone rétrécie. Ce test est utilisé chez les nouveau-nés qui sont incapables d’évacuer les selles. Pour cela le médecin introduit dans le côlon un liquide contenant du baryum, via l'anus. Ceci permet d'observer l'intestin aux rayons X. Ce test est aussi utile chez les enfants afin de déterminer sur quelle longueur le côlon est affecté.
Chez l'enfant ou l'adulte un test de manométrie est également possible : un petit ballon est introduit dans le rectum du patient pour mesurer la pression des muscles du sphincter de l’anus et déterminer à quel point la personne est capable de ressentir les différentes sensations de plénitude dans le rectum. Chez un patient atteint de la maladie de Hirschprung les muscles du rectum ne se détendent pas normalement.
Le diagnostic repose essentiellement sur la biopsie intestinale du rectum. Le médecin prélève un fragment du rectum et l’examine ; en l'absence de cellule ganglionnaire et en cas d'hypertrophie des fibres nerveuses parasympathiques la maladie de Hirschsprung est diagnostiquée.
Diagnostic différentiel
Il vise à éviter une éventuelle confusion avec d’autres pathologies possibles et aux mêmes symptômes, telles que :
- la pseudo-obstruction intestinale chronique (POIC), un accident sub-occlusif à répétition qui peut être d’origine myopathique ou neuropathique ;
- le syndrome du bouchon méconial, dans le cas où les bilans Hirschsprung et de la mucoviscidose sont négatifs (test de la sueur, et biopsies). Il peut être caractérisé par une amélioration spontanée ;
- le micro-côlon gauche, fréquent chez de gros bébés et bébés nés de mère diabétique.
Causes
C’est une maladie polygénique, c’est-à-dire due à la mutation de plusieurs gènes indépendants, dont une combinaison particulière détermine cette malformation.
Formes non syndromiques
Plusieurs gènes ont été identifiés comme responsables de maladie de Hirschsprung :
- RET situé sur le locus q11.2 du chromosome10,gène le plus souvent impliqué dans les formes familiales de maladie de Hirschsprung[2].(en) 164761 (rearranged during transfection protooncogene) ;
- GDNF situé sur le locus p13.1-p12 du chromosome 5 ;
- NRTN situé sur le locus p13.3 du chromosome 19 ;
- EDNRB situé sur le locus q22 du chromosome 13 ;
- EDN3 situé sur le locus q13.2-q13.3 du chromosome 20 ;
- ECE1 situé sur le locus p36.1 du chromosome 1.
Toutefois, aucune anomalie génétique n'est retrouvée dans un cas sur deux[3].
Formes génétiques syndromiques
| Syndromes | Signes cliniques | Transmission | Chromosome locus |
Gène | Fréquence | Numéro MIM |
|---|---|---|---|---|---|---|
| Syndrome de Bardet-Biedl | Retard mental Obésité Polydactylie Rétinite pigmentaire Anomalies rénales et génitales |
Récessive | 7 locus connus | 2-10 % | 209900 | |
| Chondrodysplasie métaphysaire autosomique récessive | Nanisme Cheveux rares Déficit immunitaire |
Récessive | 9 p13 | RMRP | 7-9 % | 250250 |
| Syndrome d'Ondine | Hypoventilation Neuroblastome |
Dominante | PHOX2B | 16 % | 209880 | |
| Dysautonomie familiale | Récessive | 9 q31 | IKBKAP | ? | ||
| Syndrome de Mowat-Wilson | Microcéphalie Retard mental Agénésie du corps calleux Cardiopathie congénitale Épilepsie Petite taille |
Dominante | 2q22 | ZFHX1B | 62-70 % | 235730 |
| Pseudo-obstruction intestinale chronique idiopathique | Dominante et Récessive | ? | ? | Jusque 20 % | 243180 | |
| Syndrome de Goldberg-Shprintzen | Récessive | 10 q21.1 | KIAA1279 | 609460 | ||
| Néoplasie endocrinienne multiple type 2 | Dominante | RET | Rare | 171400 | ||
| Neurofibromatose type 1 | Dominante | Inconnu | 162200 | |||
| Syndrome de Smith-Lemli-Opitz | Retard mental Syndactylie Cardiopathie Anomalies sexuelles |
Récessive | 11 q12-q13 | DHCR7 | Inconnu | 270400 |
| Syndrome de Waardenburg-Shah | Dominante et Récessive | Presque 100 % | ||||
| Syndrome L1 | Retard mental Agénésie du corps calleux Hydrocéphalie Pouce en adduction |
Récessive à l'X | X | L1CAM | Inconnu |
- SOX10 dans le syndrome Yemenite 4 situé sur le locus q13.1 du chromosome22
Chromosomique
- Trisomie 21, probablement par l'intermédiaire du gène DSCAM[4]
- Délétion 10q
- Délétion 13q
- Délétion 2q22
Cause inconnue
C'est le cas pour 10 à 15 % des cas .
Une étude cas-témoins américaine publiée en 2016 montre que cette malformation et d'autres survenant également en début de grossesse à un moment particulier de l'embryogenèse (ce qui laisse suspecter une cause environnementale telle que l'exposition de la mère à un produit tératogène et/ou perturbateur endocrinien à ce moment) ; cette étude faite en Caroline du Nord a conclu à une association significative entre le fait de résider pour la mère près d'une zone agricole utilisant des pesticides et certaines malformations et cette maladie ou d'autres « en particulier, les atrésies de l'œsophage »[5]. Ces types de malformations semblent souvent pouvoir être associé à des facteurs environnementaux[6].
Traitement
Le traitement de la maladie de Hirschsprung est un traitement chirurgical. La zone malade est enlevée et la continuité digestive va être rétablie. Deux traitements sont possibles : une prise en charge immédiate, qui va être effectuée le plus tôt possible, et une prise en charge secondaire, qui est le traitement définitif.
Lors de la prise en charge immédiate, le diagnostic est fait lors des premiers jours de la vie du nouveau-né. Une fois que le diagnostic est établi, le bébé va être pris en charge par un service pédiatrique. Le premier traitement va être le nursing, il consiste à introduire une sonde dans le rectum ou à effectuer de petits lavements prudents. Ceci a pour but de permettre l'évacuation des selles et le déballonnage de l’enfant. Le nursing n’est pas sans risque, il existe des risques de perforations. Cette technique peut marcher. Dans ce cas, l’enfant va être réalimenté et il va rentrer chez lui. Ses parents devront lui faire des soins de nursing. Si après son retour, le nouveau-né a de la fièvre, des selles liquides ou des vomissements, il doit revenir à l’hôpital. Si l’enfant n’est pas bien pris en charge, il risque une entérocolite aiguë.
Si le nursing ne fonctionne pas, le nouveau-né sera pris en charge et on lui fera une chirurgie de décompression, c’est une colostomie. Elle consiste à effectuer une ouverture chirurgicale de la paroi du côlon permettant ainsi son exploration. Elle va permettre de faire une biopsie et d’analyser son innervation. Elle permet aussi la décompression et l’évacuation du côlon. Une fois l’opération réalisée, l’enfant va être réalimenté progressivement et on va surveiller que les selles ne soient pas trop liquides ou trop importantes avant de le laisser sortir. Si la maladie de Hirschsprung est très étendue, la réalimentation se fera par cathéter.
Le traitement secondaire va être le traitement définitif. L’enfant est mis sous traitement antibiotique avant l’opération et il le continuera 48 heures après qu’elle sera passée. Ce traitement permet de prévenir toute infection, car c’est la plus grosse complication possible. Elle peut être due à l’entérocolite ou à une contamination par les selles. Cette chirurgie peut être pratiquée avant les 3 premiers mois de vie si nécessaire. Il existe différentes techniques pour ce traitement mais toutes ces opérations ont le même but. Elles permettent de rétablir la continuité du tube digestif après avoir effectué une ablation partielle de la partie du côlon malade. Après cette ablation de la portion pathologique du côlon, le segment de l'iléon est relié au segment du côlon qui reste, avec du fil ou des agrafes. Cette intervention n'entraîne généralement pas de conséquences sur le fonctionnement du tube digestif. Quand l'ensemble du côlon est atteint, c'est l'iléon normalement innervé qui doit être amené au niveau du rectum ou même de l'anus. Autrement dit, le but recherché est de supprimer les zones intestinales ne contenant plus de cellules neuro-ganglionnaires, et de relier les intestins qui fonctionnent normalement à la partie terminale du tube digestif, c'est-à-dire le rectum, si celui-ci possède ces cellules, sinon à l'anus. Une technique alternative consiste en une résection transanale du colon distal avec des résultats comparables voire supérieurs à la technique classique[7].
Cette opération est effectuée de plus en plus tôt, dans les premières semaines de vie de l’enfant afin de ne pas opérer ces enfants alors que l’intestin est déjà trop dilaté. Cela permet d’éviter des dysfonctionnements.
Une fois l’opération terminée, l’enfant gardera une sonde urinaire 48h. Suivant la technique utilisée, l’enfant peut développer des fistules anastomotiques ou un abcès local, par exemple à la suite de l’opération. Après un certain temps, on peut observer des troubles dans les selles : constipations, diarrhée, incontinence. Cependant, ces troubles sont observés sur une infime partie des personnes ayant subi cette opération. De plus, dans les cas d’incontinence ou de souillures, ils s'améliorent avec une rééducation périnéale. Il y a aussi un risque de mortalité même s'il est faible.
Il est important de faire garder ensuite à l'enfant un régime alimentaire à forte teneur en fibres et liquides, afin d'éviter la constipation.
Notes et références
- ↑ https://www.pourlascience.fr/sd/medecine/le-mystere-hirschsprung-10072.php
- 1 2 Amiel J, Sproat-Emison E, Garcia-Barcelo M et al. Hirschsprung disease, associated syndromes and genetics: a review, J Med Genet, 2008;45:1–14
- ↑ Butler Tjaden NE, Trainor PA, The developmental etiology and pathogenesis of Hirschsprung disease, Transl Res, 2013;356:1-15
- ↑ Jannot AS, Pelet A, Henrion-Caude A et al. Chromosome 21 scan in Down syndrome reveals DSCAM as a predisposing locus in Hirschsprung disease, PLoS One, 2013;8:e62519
- ↑ Rappazzo, K. M., Warren, J. L., Meyer, R. E., Herring, A. H., Sanders, A. P., Brownstein, N. C., & Luben, T. J. (2016). Maternal residential exposure to agricultural pesticides and birth defects in a 2003 to 2005 North Carolina birth cohort. Birth Defects Research Part A: Clinical and Molecular Teratology, 106(4), 240-249 et illustration/schéma .
- ↑ Amar E (2017). [ Malformations et facteurs environnementaux]. Revue de médecine périnatale, 9(2), 73-80 |accès à l'étude.
- ↑ Chen Y, Nah SA, Laksmi NK et al. Transanal endorectal pull-through versus transabdominal approach for Hirschsprung’s disease: a systematic review and meta-analysis, J Pediatr Surg, 2013;356:642-51
Voir aussi
Sources
- (en) Online Mendelian Inheritance in Man, OMIM (TM). Johns Hopkins University, Baltimore, MD. MIM Number: 142623
- (en) Melissa A Parisi, Hirschsprung Disease Overview dans GeneReviews at GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1997-2005.
- North American Society for Pediatric Gastroenterology, Hepatology and Nutrition : La maladie de Hirschsprung.
Liens externes
Bibliographie
- Núñez-Ramos, R., Fernández, R. M., González-Velasco, M., Ruiz-Contreras, J., Galán-Gómez, E., Núñez-Núñez, R., & Borrego, S. (2016). A Scoring System to Predict the Severity of Hirschsprung Disease at Diagnosis and its Correlation with Molecular Genetics. Pediatric and Developmental Pathology (résumé).