
| Spécialité | Génétique médicale et neurologie |
|---|
| CISP-2 | A90 |
|---|---|
| CIM-10 | Q90 |
| CIM-9 | 758.0 |
| OMIM | 190685 |
| DiseasesDB | 3898 |
| MedlinePlus | 000997 |
| eMedicine | 943216 |
| MeSH | D004314 |
| Patient UK | Downs-syndrome-trisomy-21 |
![]() Mise en garde médicale
Mise en garde médicale
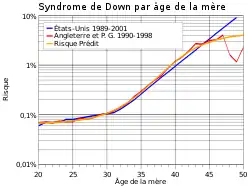
La trisomie 21 (ou syndrome de Down) est une anomalie chromosomique congénitale provoquée par la présence d'un chromosome surnuméraire pour la 21e paire. Ses signes cliniques sont très nets, un retard cognitif est observé, associé à des modifications morphologiques particulières. C'est l'une des anomalies génétiques les plus communes aux États-Unis en 1996, avec une prévalence de 9,2 pour 10 000 naissances vivantes[1]. L’incidence est d'environ 1 pour 770 naissances[2], toutes grossesses confondues, et varie en fonction de l'âge de la mère (seul facteur de risque connu à ce jour) : environ 1/1 500 à 20 ans, 1/900 à 30 ans et 1/100 à 40 ans[3]. Cette anomalie ne doit pas être confondue avec d'autres trisomies comme le syndrome XYY ou l'anomalie XXY qui ont des manifestations cliniques très différentes.
L'un des traits les plus notables du syndrome de Down est le déficit du développement cognitif, mais on constate aussi dans ce cas des malformations congénitales comme des cardiopathies[4]. Le QI des enfants atteints de trisomie 21 est extrêmement variable. Un certain nombre de patients souffrent de complications dites « orthopédiques » imposant l'hospitalisation[5]. Les anomalies musculo-squelettiques sont souvent source de complications. Avec les progrès de la médecine et le suivi paramédical (telle que l’orthophonie), la qualité de vie des personnes trisomiques 21 s’est considérablement améliorée, ainsi que leur espérance de vie.
La trisomie 21 a également été appelée mongolisme mais ce terme, bien qu'encore utilisé familièrement, est considéré aujourd'hui comme désuet et péjoratif.
Épidémiologie
La trisomie 21 est l'explication la plus fréquente de retard mental, à l'origine de 25 % des handicaps mentaux chez les enfants d’âge scolaire[6].
Parmi les anomalies chromosomiques observées en cours de grossesse, il s’agit de celle dont l’incidence est la plus élevée : aux alentours de 1/770 naissances, soit 1,3/1 000 naissances. Cette incidence s’accroît avec l'âge de la mère. Actuellement, avec l'utilisation des techniques modernes de dépistage, la proportion a été portée à 1/2 000 naissances. En France, 65 000 à 70 000 personnes atteintes de la trisomie 21 sont recensées[7]. Néanmoins, la prévalence tend à s’accroître, en raison de l'allongement de vie des trisomiques[8].
Il s'agit d'une cause fréquente d'interruption médicale de grossesse : aux États-Unis, elle concerne entre 50 et 85 % des cas de trisomie diagnostiquée avant la naissance[9].
Clinique
On estime que, hors avortement provoqué, la gestation d'un fœtus atteint par la trisomie 21 se terminera par une fausse couche spontanée ou une mortinatalité dans 75% des cas[10].
Manifestations cognitives
La trisomie 21 libre complète et homogène, la forme la plus fréquente, s'accompagne habituellement d'une déficience intellectuelle variable, souvent légère[11], le quotient intellectuel moyen chez les jeunes adultes est de 50, équivalent à celui d'un enfant de 8–9 ans[12], et il faut insister sur les points suivants :
- l'importance de cette déficience est très variable d'une personne à l'autre, au même titre que les capacités intellectuelles des individus sains. Certaines personnes trisomiques 21 atteignant l'âge adulte savent lire et écrire avec aisance et ont une autonomie pratiquement complète quand d'autres ont une faible autonomie ;
- il n'y a pas de lien direct entre la rapidité du développement psychomoteur du jeune enfant et ses performances à l'âge adulte ;
- il est très difficile d'évaluer les capacités intellectuelles d'une personne trisomique 21, car la plupart des épreuves psychométriques requièrent des capacités de coordination motrice ou visuelle, de tonus musculaire, de langage ou de communication qui font que les capacités intellectuelles des personnes trisomiques 21, souvent peu performantes dans ces domaines, sont fréquemment sous-évaluées.
Pour les trisomies en mosaïque, la situation est plus complexe car la proportion de cellules trisomiques 21 dans le cerveau n'est pas connue. Dans quelques rares cas de trisomie 21 dérivée d'une translocation, les capacités intellectuelles peuvent être normales.
Manifestations physiques
Les signes de la trisomie 21 changent avec l'âge.
Le signe le plus fréquent est l'hypotonie musculaire globale associée à une hyperlaxité des ligaments.
Certains signes physiques permettent souvent le diagnostic de la trisomie 21 :
- la tête est petite et ronde avec un visage plutôt aplati et une nuque plate. Les fentes des paupières sont obliques, en haut et en dehors, les yeux sont très écartés. Il y a souvent un strabisme ou un nystagmus. Les iris, lorsqu'ils sont clairs, peuvent avoir des taches blanches caractéristiques, dites de Brushfield. La racine du nez est peu marquée en raison du moindre développement des os du nez et s'accompagne d'un épicanthus, repli cutané formant comme une troisième paupière. Les pavillons des oreilles sont petits et mous, avec des conduits auditifs souvent étroits. Le palais est parfois ogival et la langue peut sortir de la bouche (en position de repos, la langue est normalement collée au palais qui se développe sur elle, chez les trisomiques 21, elle est en position basse ce qui entraîne une insuffisance de développement du palais) ;
- le cou est parfois court et large, le thorax déformé, l'abdomen mou avec un écart des muscles abdominaux grands droits, source de hernie ombilicale, le pénis est souvent petit avec cryptorchidie ;
- les mains sont souvent trapues, avec une inclinaison du 5e doigt vers l'intérieur. Les doigts sont courts car les phalanges du milieu y sont trop courtes (brachymésophalangie). Dans la paume de la main, les plis peuvent être horizontaux, il existe souvent un seul pli transverse. L'examen à la loupe des dermatoglyphes (petits reliefs cutanés présents sur la pulpe des doigts et sur les paumes) montre aussi des figures spécifiques ;
- les pieds sont, eux aussi, courts, avec un grand espace entre les deux premiers orteils, et assez souvent une mauvaise implantation d'un ou plusieurs orteils. Il peut exister des syndactylies (fusion de deux doigts ou orteils) aux mains ou aux pieds.
À la naissance, le médecin recherche systématiquement certains types de malformations :
- cardiaques (jusqu'à 50 % des nouveau-nés trisomiques) : communications inter-auriculaires, inter-ventriculaires ou auriculo-ventriculaires [13];
- oculaires : cataracte ou glaucome congénitaux ;
- digestives : sténose duodénale (rétrécissement du duodénum au niveau de l'intestin) ou maladie d'Hirschsprung (chez 2 % des trisomiques, tandis que 12 % des patients atteints de Hirschsprung sont trisomiques)[13].
- orthopédiques : au niveau des hanches et des vertèbres.
Diagnostic

La trisomie 21 est une anomalie chromosomique définie par la présence d'un 3e exemplaire plus ou moins complet du chromosome 21. Dans 95 % des cas, il s'agit d'une trisomie 21 dite « libre » (par non-disjonction méiotique) et homogène. Dans 2 à 3 % des cas, il s'agit d'une mosaïque. Enfin, dans 2 à 3 % des cas restants, il s'agit d'une trisomie dite « non libre », c'est-à-dire que la partie surnuméraire du chromosome 21 est fusionnée avec un autre chromosome[14].
Le diagnostic ne peut se faire que par la mise en évidence du chromosome 21 supplémentaire lors d'un examen génétique, généralement par analyse du caryotype ou par hybridation in situ en fluorescence.
Trisomie libre
La présence d'un troisième chromosome 21 est la cause de la pathologie. Le mécanisme de la présence du chromosome supplémentaire est important à connaître pour le conseil génétique. La réalisation du caryotype permet de connaître le mécanisme.
La formule chromosomique de la personne atteinte de trisomie 21 est donc : « 47, 21+ ».
Le chromosome 21 supplémentaire vient presque toujours de la mère. L'origine de cette maladie génétique se situe lors de la gamétogénèse, et plus précisément lors de la répartition des chromosomes homologues au cours de la méiose.
-- Lors de l'anaphase 1 : les chromosomes homologues de la paire no 21 appariés en bivalent ne se sont pas séparés et ont migré vers un même pôle.
-- Lors de l'anaphase 2 : les deux chromatides du chromosome 21 ont migré vers un même pôle après clivage du centromère. Donnant ainsi un trisome et un monosome. Sachant que le monosome n'est pas viable. (sauf dans le cas du syndrome de Turner 45,X)
Trisomie non libre
Il s'agit de la fusion de deux chromosomes 21 par le mécanisme dit de translocation. Il s'agit donc d'un chromosome apparent ayant le contenu génétique de deux chromosomes. La formule chromosomique de la personne atteinte de cette forme de trisomie 21 est donc « 46,XY,+21,der(21)t(21;21) » ou « 46,XX,+21,der(21)t(21;21) ».
La trisomie 21 par translocation est dans certains cas héritée de l'un des parents. Il faut dans ce cas pratiquer un caryotype chez les parents pour identifier le porteur de l'anomalie.
Pronostic et prise en charge
Diverses conditions pathologiques peuvent survenir ou s'associer à la trisomie 21, plus fréquemment qu'en population générale. Quelques-unes nécessitent d'être traitées rapidement dès la naissance, les autres une surveillance médicale tout au long de la vie[15].
La prise en charge médicale est multidisciplinaire, adaptée à chaque personne. Le suivi médical doit tenir compte du fait que la personne trisomique analyse moins ses sensations corporelles, qu'elle les exprime moins, et ne se plaint guère[16]. L'éducation des parents, informés des complications possibles, est essentielle pour détecter l'apparition de ces troubles[13].
Les progrès de la prise en charge médicosociale ont permis une augmentation importante de l'espérance de vie, de l'ordre de 30 ans en 1973 jusqu'à 60 ans en 2002. Celle-ci continue d'augmenter mais en restant inférieure à celle de la population générale. Une bonne prise en charge peut aider une personne porteuse de trisomie 21 à mener une vie pleine et productive[13].
Complications cardiovasculaires et pulmonaires
Une cardiopathie congénitale est retrouvée dans la moitié des cas, essentiellement à type de communications interauriculaires ou interventriculaires[17]. À la naissance, le pronostic dépend essentiellement de la réparation des malformations cardiaques associées. Les progrès de la chirurgie cardiaque ont contribué à une espérance de vie qui dépasse désormais les cinquante ans[8].
La maladie de moyamoya est une anomalie rare, mais qui se retrouve parmi les trisomiques, en pouvant bénéficier d'une chirurgie vasculaire[15].
La cause des décès est surtout cardiaque et pulmonaire[8], complications liées à une éventuelle hypertension pulmonaire (1 à 5% des personnes) ou au rétrécissement des voies respiratoires[15].
Croissance et problèmes orthopédiques
La croissance se déroule de la même façon qu'en population générale, mais avec des niveaux de 2 à 3 déviations standard inférieurs à la moyenne. Par exemple, la taille finale est proche de 160 cm pour les hommes et de 145 cm pour les femmes.
L'obésité est fréquente[18], survenant chez 25 % des enfants et 50 % des adultes, ce qui favorise les apnées de sommeil, le diabète, les troubles cardio-pulmonaires[15]. Ces troubles peuvent être prévenus et améliorés par une bonne diététique, l'exercice physique et une prise en charge précoce de l'hypotonie bucco-faciale et des troubles de la mastication[16].
Les troubles squelettiques sont habituels avec troubles de la marche ou de la préhension. Ils sont liés à une hypotonie et hyperlaxité qui augmentent le risque de luxation (hanche, rotule…) ou à des pieds plats[16]. La condition la plus dangereuse est l'instabilité des premières vertèbres cervicales (articulation atlanto-axoidienne) avec un risque de compression médullaire[15] ; cette situation est plutôt rare (moins de 2 % des enfants trisomiques), mais elle peut limiter une pratique sportive[16],[19].
Il existe un risque accru d'ostéoporose[20] du à plusieurs facteurs, dont un déficit en vitamine D par défaut d'exposition solaire ou trouble de l'absorption intestinale[13].
Troubles digestifs
Les éventuelles malformations digestives, qui peuvent survenir de la bouche à l'anus, sont détectées à la naissance. Les enfants trisomiques sont plus susceptibles que les autres de présenter un reflux gastro-oesophagien, une constipation chronique, une diarrhée intermittente. Une maladie cœliaque existe chez 5 % d'entre eux, un dépistage est recommandé et si le diagnostic est posé, un régime sans gluten doit être suivi tout au long de la vie[13].
Troubles endocriniens
L'hypothyroïdie se voit jusqu'à un tiers des cas[8]. Elle peut être congénitale ou acquise au cours de la vie, ce qui nécessite un suivi régulier en sus du dépistage à la naissance[13].
La puberté apparait le plus souvent à l'âge habituel, mais elle peut être retardée dans les deux sexes. Chez les garçons, il y a en général des troubles testiculaires, comme une cryptorchidie ou une oligospermie, avec stérilité[13] (seuls deux cas de paternité ont été publiés à la date de 2004)[21].
Chez les jeunes filles, la fertilité est préservée, avec un risque de transmettre l'anomalie à la descendance de 25 à 50 %[8],[16]. Une contraception adaptée est proposée[16].
Par rapport à la population générale, la ménopause survient en moyenne quatre ans plus tôt[21]. Le risque de cancer gynécologique est moindre chez les femmes, mais chez les garçons le risque de cancer du testicule est augmenté[16].
Troubles hématologiques
Des troubles transitoires, comme une réaction myéloblastique, peuvent survenir de façon transitoire dans les trois premières années de vie (10 % des sujets). Si ce trouble apparait au cours de la vie fœtale, il peut entrainer un avortement spontané[13],[16].
Le risque de leucémie (autour de 2 %) est beaucoup plus élevé qu'en population générale, il s'agit le plus souvent de leucémie aigüe lymphoblastique, plus rarement de leucémie aigüe myéloïde. Le pic de fréquence se situe autour de l'âge de 3 à 5 ans, avec une bonne réponse à la chimiothérapie[15],[16], et un autre vers l'âge de 30 ans[22].
Un déficit en fer est fréquent[15].
Immunité et infections
Il n'y a pas de déficit vraiment important de l'immunité chez les patients trisomiques 21, quoiqu'un déficit en Ig A soit courant. La plus grande fréquence des infections ORL, respiratoires et cutanées est liée à d'autres causes (reflux gastro-œsophagien, carence en fer, malformations locales...)[16],[23]. Les infections doivent être reconnues et traitées rapidement[19].
Les troubles auto-immuns sont plus fréquents qu'en population générale : maladies de la thyroïde, diabète, maladie cœliaque... déjà citées ainsi que des maladies articulaires comme l'arthrite chronique juvénile, et des maladies cutanées comme la pelade, le vitiligo ou le psoriasis[15].
La trisomie 21 n'est pas une contre-indication à la vaccination, et le calendrier vaccinal recommandé doit être suivi[19].
Troubles auditifs et visuels
Les infections ORL (otites) et des anomalies de l'oreille interne favorisent une perte d'audition (hypoacousie) chez les patients trisomiques 21, ce qui peut entrainer un retard d'acquisition du langage. La détection et le traitement précoces de ces troubles permettent un meilleur développement intellectuel[13],[15].
Les problèmes visuels sont habituels. Une cataracte congénitale peut se manifester à la naissance ou se développer dans le jeune âge. Les troubles les plus fréquents sont les troubles de la réfraction, surtout la myopie, puis le strabisme et le nystagmus. La surveillance et le traitement de ces anomalies sont importants pour la qualité de vie des enfants trisomiques 21[13],[15].
Une étude publiée le 1er septembre 2022 dans la revue Science montre qu'une thérapie basée sur l'injection de l'hormone GnRH chez des patients atteints de trisomie 21 aurait des effets positifs sur leurs fonctions visio-spatiales, mais aussi sur d'autres fonctions cognitives[24].
Développement intellectuel et psychomoteur
Enfance
Presque tous les enfants trisomiques 21 ont des difficultés d'apprentissage, de minimes à modérées, par surexpression de certains gènes liée à la trisomie[13]. Cependant, la qualité du développement intellectuel de l'enfant trisomique 21 dépend en grande partie de la qualité de la prise en charge éducative précoce[15],[25].
Les étapes du développement psychomoteur sont retardées (marche acquise chez 50 % des enfants après 24 mois[26], babillage vers 15 mois en moyenne[27], assemblage de deux mots entre 2 et 5 ans). L'acquisition du vocabulaire est riche, mais celle de la grammaire est pauvre (5 % des trisomiques ne dépassent pas le stade des mots uniques). Le niveau de compréhension, notamment l'apprentissage visuel, est bien meilleur que les capacités d'expression (difficultés d'élocution)[19],[25].
Plus de deux enfants trisomiques sur trois peuvent acquérir des notions de lecture et d'écriture, alors que le calcul reste un point faible. Le quotient intellectuel entre 20 et 80 (le QI moyen en population générale est de 100 avec une déviation standard de 15, considéré normal par convention entre 80 et 120). Les capacités finales de chaque enfant trisomique ne sont pas prévisibles, car liées en partie à l'apprentissage et à l'entraînement. Environ 10 % atteignent le niveau « normal faible » alors que la même proportion accusent un retard profond[25].
La plupart des enfants trisomiques bénéficient d'être maintenus à l'école primaire avec un soutien ou un encadrement spécifique, le plus souvent en classes spécialisées[16].
5 à 13 % des enfants trisomiques peuvent présenter des crises convulsives, comme le syndrome de West ou le syndrome de Lenox-Gastaut. Par rapport à la population générale le risque d'épilepsie est accru, il peut survenir à l'âge adulte avec une bonne réponse aux traitements[13].
7 à 16 % ont des troubles autistiques et environ 6 % un trouble de déficit de l'attention, ce qui nécessite un dépistage approprié et une prise en charge adaptée (thérapies comportementales, médications....)[15],[19]. À un âge plus avancé, de 11 à 14 ans, un trouble rare mais grave peut survenir, le trouble désintégratif de l'enfance.
Les personnes trisomiques sont conscientes de leur handicap et peuvent en souffrir, avec des troubles dépressifs et anxieux plus fréquents. Elles sont plus sensibles aux stress emotionnels[19].
Vie adulte
La majorité des trisomiques adultes peuvent avoir une vie semi-indépendante et autonome en milieu favorable, notamment une autonomie hygiénique, mais la plupart d'entre eux ne savent gérer leur argent, se déplacer seuls en milieu non familier ou répondre à des situations imprévues. Les adolescents et adultes trisomiques tendent à être anxieux, introvertis, solitaires et passifs[25].
Des adultes trisomiques peuvent travailler en milieu professionnel protégé et certains sont devenus des professionnels à succès, par exemple artistes ou acteurs[19]. Ils ont la capacité de mener une vie affective normale et peuvent être sexuellement actifs[25]. Au Danemark, 1 à 2% de personnes trisomiques sont mariées ou ont un enfant[28].
La ménopause survient en moyenne quatre ans plus tôt qu'en population générale[21]. Après l'âge de 50 ans, il existe un risque plus fréquent de démence, notamment de maladie d'Alhzeimer, avec un déclin cognitif plus précoce vers l'âge de 60 ans[15]. C'est une cause principale de décès des trisomiques âgés[19].
Dépistage prénatal
Le diagnostic prénatal est possible, il s'effectue soit au premier trimestre, à partir d'un prélèvement de villosités choriales ou choriocentèse qui est fait à partir de 11 semaines jusqu'à 14 semaines, soit par ponction de liquide amniotique au-delà de 14 semaines. Le risque d'avortement iatrogène est plus élevé lors d'une choriocentèse (environ 1 %) et le choix du prélèvement est fait en fonction du degré d'urgence et de l'indication médicale. Lorsque l'âge gestationnel est très avancé (vers la 30e semaine) un prélèvement de sang fœtal en ponctionnant le cordon ombilical peut être réalisé. Il permet d'obtenir un résultat rapidement car la culture de cellules sanguines (généralement de lymphocytes) ne demande que quelques jours alors qu'il faut plusieurs semaines pour les amniocytes.
La recherche d'ADN fœtal circulant dans la circulation maternelle est techniquement possible mais onéreuse, elle est remboursée depuis janvier 2019 par l'assurance maladie française ; c'est le dépistage non-invasif de la trisomie 21[29], ou TGNI[30], aussi appelé DPNI. L'étude SEHDA (séquençage à haut débit des aneuploïdies) confirme une sensibilité (99,79 % chez les femmes à risque modéré) et une spécificité du test supérieures à 99 % : il permet d'éviter 95 % des gestes invasifs, soit amniocentèse, soit biopsie[31].
Plusieurs études conduites aux États-Unis ou au Royaume-Uni ont montré que 90 à 93 % des grossesses ayant donné lieu au diagnostic de trisomie 21 ont été interrompues[32],[33]. En France, le pourcentage d’interruption de grossesse à la suite d'un diagnostic prénatal de trisomie 21 est de 96 %[34].
Selon une étude de 2012 de l'université de Caroline du Sud, les deux tiers des femmes enceintes découvrant que leur fœtus présente le syndrome de Down décident d'avorter. Cette proportion varie selon les milieux, en fonction des croyances religieuses, du lieu de vie ou encore du niveau de revenu. La chercheuse Laura Hercher remarque que « L'accès aux tests prénataux a ainsi modifié l'incidence de la trisomie 21 au point d'en faire un marqueur géographique et de classe. Comme les familles aisées ont de moins en moins d'enfants qui en sont atteints, il est de plus en plus associé à d'autres milieux[35]. »
Évaluation
Le principe de l'évaluation consiste à évaluer la probabilité de trisomie 21 afin de décider si le risque de la ponction est tolérable. Le but de l'évaluation du risque est, lorsqu'il est élevé, de permettre aux futurs parents de décider de pratiquer une amniocentèse, qui permettra le diagnostic. Le seuil de tolérance est un choix arbitraire, il est en effet difficile de comparer la possibilité de faire éliminer un fœtus sain avec celle de donner naissance à un enfant trisomique.
Le but des examens de dépistage est, en fonction d’un ou de plusieurs paramètres cliniques ou paracliniques, de séparer les femmes enceintes en deux sous-populations : une population dont le risque est jugé bas et pour laquelle il doit être abstenu de réaliser tout prélèvement invasif en expliquant néanmoins que l'anomalie peut être présente, et une population estimée à haut risque pour laquelle un examen invasif va être proposé au couple après information concernant le risque lié à la méthode de diagnostic employée. L'évaluation d'un risque fait appel aux méthodes statistiques et de probabilité suivantes : avoir des signes pertinents, c'est-à-dire connaître la sensibilité, la spécificité, la valeur prédictive positive et négative et enfin la prévalence du signe. Calculer la probabilité individuelle d'un patient d'avoir une pathologie donnée après passage d'un test de dépistage et en partant d'un risque initial déterminé dans la population à laquelle appartient ce patient : utilisation du théorème de Bayes.
Ce tableau inspiré de l'article paru dans le British Medical Journal résume l'ensemble des signes disponibles dans le cadre d'un dépistage de la trisomie 21. Les marqueurs en italique sont en cours d'évaluation[36].
| Différents marqueurs pour le dépistage de la trisomie 21 | |
|---|---|
| Marqueurs | Risque augmenté si |
| Marqueur clinique | |
| Âge | Élevé |
| Marqueurs sériques au premier trimestre | |
| Bêta-hCG libre | Élevée |
| PAPP-A (en) | Bas |
| ADAM12 | Basse |
| Marqueurs sériques au deuxième trimestre | |
| Alpha-fœtoprotéine | Basse |
| Estriol libre | Bas |
| HCG totale | Élevée |
| Inhibine | Élevée |
| Marqueurs échographiques | |
| Épaisseur de la clarté nucale | Élevée |
| Os propres du nez | Absents |
| Index de pulsatilité du ductus venosus | Élevé |
| Régurgitation de la valve tricuspide | Présente |
Au premier trimestre
Échographie
Cette technique d'évaluation de la trisomie 21 (et des autres aneuploïdies) doit beaucoup au Professeur Kýpros Nicolaïdis de la Fetal Medicine Foundation de Londres.
Le marqueur échographique utilisé est la clarté nucale. La technique de mesure doit répondre à des critères stricts.
Il existe un autre marqueur échographique mais de mesure plus délicate : l'os propre du nez.

Biologie
Les substances utilisées sont l'hCG libre et la PAPP-A (Pregnancy-associated plasma protein A (en)).
Le dépistage par ces marqueurs sériques se fait entre les 11e et 14e semaines d’aménorrhées sur une simple prise de sang.
Les facteurs pris en compte pour le calcul du risque de trisomie 21 sont l'âge maternel, le terme de la grossesse, le poids et le tabagisme de la future maman ainsi que les antécédents de trisomie 21 dans la famille. Dernier marqueur indispensable : la clarté nucale, qui est mesurée lors de l'échographie du 1er trimestre.
De nouveaux tests sanguins basés sur l'analyse de l'ADN fœtal dans le sang de la mère ont récemment été évalués par la Haute Autorité de santé comme permettant de dépister 99 % des trisomies 21 chez les « femmes à risques » dès la cinquième semaine de grossesse et permettrait ainsi d'éviter 180 à 360 fausses couches par an en France[37].
Au second trimestre
Échographie
Aucun signe échographique n'est symptomatique de la trisomie 21. Néanmoins, un certain nombre d'anomalies mineures ou majeures qui se rencontrent plus fréquemment dans cette maladie chromosomique peuvent être mis en évidence.
- Anomalies mineures
- hypoplasie ou absence des os propres du nez ;
- fémur court, inférieur au 5e centile pour l'âge ;
- épaisseur de nuque supérieure à 6 mm à 20 semaines ;
- écartement important entre les premier et deuxième orteils ;
- brièveté de la deuxième phalange du cinquième doigt (brachymésophalangie) ;
- langue protruse…
- Anomalies majeures
- des malformations cardiaques (canal atrio-ventriculaire et en particulier) (dans 40 % des cas) ;
- des sténoses digestives (dans 10 à 18 % des cas) (image en « double bulle » de sténose duodénale).
Le dépistage du deuxième trimestre prend également en compte les mesures échographiques du 1er trimestre et ce qui est appelé un « risque séquentiel intégré » doit être calculé. Comme pour le dépistage du premier trimestre, celui-ci permet de limiter le nombre d'amniocentèses dû à l'intégration de la clarté nucale dans le calcul.
Biologie
L'évaluation se fait entre « 14+0 » semaines d'aménorrhée et « 17+6 » semaines d'aménorrhée. Les marqueurs utilisés sont l'alpha-fœtoprotéine et l'hCG libre. Certains laboratoires ajoutent l'estriol plasmatique mais le recours à ce troisième marqueur n'augmente pas la sensibilité et il est de fait peu fréquent. Cette technique d'évaluation par trois marqueurs sériques se nomme le triple test. Certains laboratoires pondèrent le résultat en fonction du poids de la patiente et/ou du nombre de cigarettes fumées par jour ainsi qu'avec les antécédents de trisomie 21 s'il y en a dans la famille proche (père, mère, frères et sœurs des parents et grands-parents).
Calcul
La probabilité d'avoir un enfant T21 est basée sur un calcul qui est a priori fonction de l'âge gestationnel et de l'âge de la mère. Cette probabilité diminue pendant la grossesse, car environ 30 % des fœtus avec trisomie 21 décèdent in utero.
Avant le test, une femme enceinte peut avoir un fœtus avec trisomie 21 : c'est la probabilité pré-test. Après le test, une femme peut aussi avoir un fœtus avec trisomie 21 : c'est la probabilité post-test. Le rapport de vraisemblance ou likehood ratio est le nombre multiplicateur qui permet de passer de la probabilité pré-test à la probabilité post-test. Si un signe est présent parmi 50 % des fœtus trisomiques et 5 % des fœtus sans T21, le rapport de vraisemblance est de 10, c'est-à-dire que la probabilité est multipliés par 10. Le rapport de vraisemblance est aussi vrai dans l'autre sens. Si un signe est présent parmi 20 % des fœtus T21 et 40 % des fœtus non T21, le rapport de vraisemblance est de 0,5 c'est-à-dire que la probabilité est divisée par deux. Il faut aussi s'assurer que les signes utilisés ne soient pas liés entre eux, c'est-à-dire que l'un ne dépende pas de l'autre. Il faut que ces signes soient statistiquement indépendants.
La probabilité donnée par la mesure de la clarté nucale et la probabilité calculée au moyen des dosages de l'hCG et de l'alpha-fœto-protéine (la prise de sang proposée en France à toutes les femmes enceintes) est l'intégration de tous les rapports de vraisemblance, qui demande l'utilisation d'un logiciel.
La probabilité à partir de laquelle un caryotype est réalisé dépend des pays : elle est de 1/300 en Angleterre et de 1/1000 en France[38].
Controverses liées à l'eugénisme
Le développement croissant des techniques de dépistage prénatal et le recours fréquent à l'interruption médicale de grossesse (96 % des fœtus trisomiques détectés par amniocentèse en France font l'objet d'un avortement) posent des questions d'ordre éthique, liées à l'eugénisme[39],[40]. Pour la juriste Catherine Bachelard-Jobard, dans le contexte de dépistage et de réduction des naissances d'enfants T21, « on est sans doute très proche de la définition de l'eugénisme »[39]. Le médecin français Jacques Milliez pose en 1999 la question de savoir si le dépistage systématique évoque « un eugénisme douteux » puisqu'il viserait à « supprimer une catégorie humaine ciblée », et signale que la moitié des fœtus détectés viennent au monde. Il ajoute qu'il existe un consensus sociétal en faveur de la décision d'interruption de grossesse, empêchant les couples de se poser la question de la pertinence de leur choix individuel[41].
Jean-Marie Le Méné, président de la Fondation Jérôme-Lejeune connue pour ses positions anti-avortement, considère ainsi que « la multiplication de tous les eugénismes individuels produit un eugénisme collectif[42]. » L’écrivain Bruno Deniel-Laurent, dans Éloge des phénomènes, un essai publié en 2014, pose aussi la question de l'eugénisme[43]. Le Collectif les Amis d'Éléonore, association de parents d’enfants porteurs de la trisomie 21, interpelle régulièrement les hommes politiques sur cette question, affirmant que les recours très fréquents à l'interruption médicale de grossesse envoient un message dévalorisant aux personnes concernées[44].
Histoire
En 1997, des archéologues grecs (équipe Diamandopoulos) décrivent dans le Journal of the History of Neuroscience une statuette découverte près de Thessalonique vieille de sept-mille ans. Celle-ci présente plusieurs caractéristiques qui évoquent le syndrome de Down, ce qui ferait d'elle la plus ancienne représentation sculptée de la maladie[45].
- Âge du fer, Rome : Equus Domitiani 2, fouille de 1909, conservée à l’antiquarium du Forum romain : squelette d'une jeune fille (121 cm, âge dentaire 13–14 ans) tuée par un coup violent à la tempe, probablement un sacrifice[46].
- 550 av. J.-C., Tauberbischofsheim, Allemagne : restes d'une femme de 18–20 ans[47].
- Règne de Jayavarman VII (1181-1220), Cambodge : plusieurs sculptures de la terrasse des éléphants à Angkor Thom représenteraient des personnes ayant le morphotype des trisomiques 21[48].
- 1475, Qilakitsoq, Groenland : momie 2 : enfant de quatre ans à quatre ans et demi présentant des anomalies osseuses fréquentes chez les trisomiques 21. F. Dunand suppose qu'il a été abandonné par eugénisme[49]. Philippe Charlier en doute, s'étonnant que l'on ait attendu si longtemps[46], la dysmorphie étant visible dès le plus jeune âge.
XIXe siècle et première moitié du XXe siècle : description clinique
La première description clinique évoquant une trisomie 21 est faite en 1838 par le médecin français Jean-Étienne Esquirol dans son ouvrage Des maladies mentales considérées sous le rapport médical, hygiénique et médico-légal[50] dans lequel il décrit certains signes cliniques observés chez des individus atteints par la trisomie 21. Il forge aussi le terme de diathèse furfuracée, en avançant le fait que la peau des trisomiques aurait une texture « farineuse ».
Le médecin français Édouard Séguin dresse également, en 1846, une description clinique de la trisomie 21 dans son Traitement moral, hygiène et éducation des idiots[51]. S'inspirant des travaux de Jean-Étienne Esquirol, il parle alors d’idiotie furfuracée pour désigner les phénomènes physiques caractéristiques d'un état particulier de retard mental.
Aux États-Unis, Séguin collabore avec Samuel Gridley Howe, pionnier en 1848 des écoles expérimentales ou centres éducatifs pour handicapés mentaux[52].
Syndrome de Down
_by_Sydney_Hodges.jpg.webp)
La première étude clinique formelle sur la trisomie 21 est celle d'un médecin aliéniste britannique, John Langdon-Down (1828-1896) dans son mémoire publié en 1866 et intitulé Observations on a ethnic classification of idiots[53] (en français : Observations sur une classification ethnique des idiots).
En s'inspirant de la classification des races de Johann Friedrich Blumenbach (1752-1840), Langdon-Down distingue chez les idiots une variété « éthiopienne », une variété « malaise », une variété « américaine » et une variété « mongolienne » (que d'autres appelleront aussi « tartare » ou « kalmouk »)[54],[55].
Cette dernière désigne en fait les individus atteints par la trisomie 21 qu'il décrit ainsi :
Un très grand nombre d'idiots congénitaux sont typiquement mongols […] Les cheveux ne sont pas noirs, comme chez les vrais Mongols, mais de couleur brune, raides et étriqués. La face est plate et large, et dénuée de proéminence. Les joues sont rondes et élargies latéralement. Les yeux sont placés en oblique, et les canthi internes sont anormalement distants l'un de l'autre. La fissure palpébrale est très étroite. Le front est plissé transversalement […] Les lèvres sont larges et épaisses avec des fissures transversales. La langue est longue, épaisse, et râpeuse. Le nez est petit. La peau a une teinte légèrement jaunâtre, déficiente en élasticité, donnant l'apparence d'être trop large pour le corps […] il ne peut y avoir aucun doute que ces caractéristiques ethniques sont le résultat d'une dégénérescence […] Le type mongolien d'idiotie représente plus de 10 pour cent des cas qui se sont présentés à moi. Ce sont toujours des idiots congénitaux, et jamais la conséquence d'accidents après la vie intra-utérine […] Ils ont une capacité considérable d'imitation […] Ils sont comiques […] Ils sont habituellement capables de parler ; le langage est simplet et indistinct, mais peut être amélioré grandement par une méthode bien dirigée de gymnastique de la langue. La faculté de coordination est anormale, mais pas si défectueuse qu'elle ne puisse être grandement renforcée.
.jpg.webp)
La description clinique de John Langdon-Down se révèle d'une grande justesse, à tel point que son nom a servi à désigner la maladie (syndrome de Down). Distinguer le mongolisme, c'est aussi pour Langon-Down, reconnaître des possibilités éducatives que l'on ne soupçonnerait pas au départ, même si leur espérance de vie était très en dessous de la moyenne de son époque[52].
Si Langdon-Down est reconnu comme le pionnier de l'éducation et des soins aux « mongoliens »[56],[57], ses conclusions médicales sont en partie fausses puisqu'il écrit aussi dans son mémoire : « Il ne peut y avoir aucun doute : ces caractéristiques ethniques sont le résultat d’une dégénérescence ». Ici, Down se réfère une théorie de la dégénérescence, influençé par les écrits de Charles Darwin, il y voit une évolution à rebours, ces patients représentant un retour à un type primitif[54].
En 1908, le docteur Adolphe Bloch commet la même erreur que John Langdon Down dans son article « Sur le mongolisme infantile dans la race blanche et sur d’autres anomalies qui sont des caractères normaux dans diverses races » publié dans Bulletins et Mémoires de la Société d’anthropologie de Paris où il cherche à établir des comparaisons entre les singularités « aberrantes » des Européens mongoliens et les caractères physiologiques normalement observés chez les individus de « race jaune » :
Les Mongoliens sont des dégénérés, au même titre que les autres idiots, c'est-à-dire que le faciès dit mongolique, comme toutes les autres anomalies, provient le plus souvent d'une maladie ancestrale, comme la tuberculose, le nervosisme, l’alcoolisme ou la syphilis, qui peut se transformer par l’hérédité et ne manifester son influence que par le trouble qu’elle apporte au développement régulier du fœtus. (cité par Bruno Deniel-Laurent dans son essai Éloge des phénomènes[58]).
Autres hypothèses
L'hypothèse raciale ou évolutive à rebours est loin de faire l'unanimité. L'un des fils de Down, Reginald Langdon-Down, lui-même médecin, suggère que les manifestations du syndrome de Down sont accidentelles et individuelles[54],[55].
Dans les années 1870 et 1880, des auteurs soulignent les similitudes entre le syndrome de Down et le crétinisme par hypothyroïdie[59]. D'autres que ces enfants sont « non finis », par la persistance de caractères anatomiques propres à telle ou telle phase du développement fœtal. Au tournant du XXe siècle, de nombreuses études sont publiées recensant l'ensemble des manifestations cliniques, notamment les troubles neuropathologiques et la fréquence des malformations cardiaques. Le lien entre le syndrome et l'âge avancé de la mère est reconnu, ainsi que le fait que ce syndrome est plus fréquent chez les derniers-nés de familles nombreuses[54].
Durant la première moitié du XXe siècle, une multitude de causes et de théories sont proposées : facteurs familiaux (syphilis maternelle, tuberculose, alcoolisme ou épilepsie chez les parents), troubles endocriniens (thyroïde, surrénales, hypophyse), toxiques (produits ou procédés contraceptifs ou abortifs), mauvaise implantation, choc émotionnel en début de grossesse etc.[54]
Origine chromosomique
Hypothèses
À partir de 1929, Raymond Turpin, chef du service de pédiatrie de l'hôpital Trousseau de Paris, réunit des observations en envisageant une origine génétique de la maladie. Chez les parents et germains de « mongoliens », il note la fréquence accrue de certains signes (langue fissurée, pli palmaire transverse…), ce qui le conduit à formuler l'hypothèse d'un gène pléiotrope en concluant à l'origine génétique de la maladie, contrairement aux tenants d'une origine embryologique[60].
En 1934, il envisage une anomalie chromosomique analogue à la mutation Bar de la drosophile, ce qui n'était pas vérifiable à l'époque, les moyens d'étude du caryotype humain n'existant pas encore[60]. Dans les années 1930, plusieurs auteurs suggèrent, de façon indépendante, des hypothèses chromosomiques comme le Néerlandais Petrus Johannes Waardenburg (en) (1932), l'américain Adrian Bleyer (1934) ou l'italien Guido Fanconi (1938) qui sont les premiers à envisager un accident de la disjonction chromosomique, mais ces suggestions passent inaperçues[61].
Publication de la découverte en 1959
En 1953 et 1956, Turpin renforce son équipe avec l'arrivée de Jérôme Lejeune, chercheur au CNRS, puis de Marthe Gautier, pédiatre experte en culture tissulaire, technique qu'elle venait d'acquérir aux États-Unis. Ces travaux de recherches sont favorisés par l'existence, dans le service de Turpin, d'une consultation spéciale pour enfants mongoliens[60].
En août 1956, le premier congrès international de génétique humaine se tient à Copenhague. Plusieurs chercheurs présentent les premiers caryotypes humains obtenus à partir de culture tissulaire, avec la détermination définitive du nombre des chromosomes humains (46 et non 48 comme on le pensait auparavant[62]). Turpin donne alors comme objectif prioritaire de recherches à son équipe, l'étude du caryotype des mongoliens[60].
En juillet 1958, l'équipe met en évidence la présence d'un chromosome surnuméraire, apparenté au plus petit des autosomes. Le , Turpin présente une communication orale devant l'Académie des sciences pour annoncer sa découverte, elle est suivie de la publication d'une note le 16 mars 1959, signée par Jérôme Lejeune, Marthe Gautier et Raymond Turpin, présentant neuf cas d'enfants mongoliens dotés de 47 chromosomes au lieu de 46[60],[63].
Cette découverte est immédiatement reconnue par la communauté scientifique avec un grand retentissement international. C'est la première anomalie chromosomique décrite chez l'homme, et c'est la première maladie pour laquelle est mise en évidence la relation entre le génotype et le phénotype. Ceci à cinq jours près, car le 31 janvier 1959, des chercheurs anglais, dont Patricia Jacobs, publient leur découverte d'une autre trisomie chromosomique, la XXY à l'origine du syndrome de Klinefelter[60]. Cependant la paternité de la découverte d'une trisomie a été sujette à controverse[64], la publication de Gautier et Lejeune ne mentionnant qu'un chromosome surnuméraire, sans préciser une trisomie particulière.
Durant l'année 1959, la trisomie mongolienne est confirmée par des chercheurs anglais, américains et suédois. En avril 1960, une commission internationale, dite de Denver, propose de classer les chromosomes humains en numéros selon leur taille et leur aspect, c'est ainsi que le numéro 21 a été attribué aux chromosomes de la trisomie mongolienne[60].
En 1961, dans son numéro du 8 avril, la revue The Lancet publie une lettre ouverte signée par 19 chercheurs et experts internationaux, dont les Français Lejeune et Turpin, déplorant que les termes mongolisme, mongolien, et mongoloïde ont des « connotations trompeuses » en devenant « embarrassants »[65],[66]. C'est en effet trompeur, car les mongoliens n'ont pas de gènes asiatiques, et c'est embarrassant vu la part croissante des chercheurs chinois et japonais dans ce domaine. D'autres appellations sont proposées, principalement syndrome de Down par les anglophones, et trisomie 21 par les francophones. Cette lettre ouverte est aussi reprise par le American Journal of Human Genetics en décembre 1961[67]. En France, le mongolisme est donc renommé par Jérôme Lejeune, « trisomie 21 », « tri » voulant dire « trois » et « some » voulant dire « chromosome », c’est-à-dire « trois chromosomes 21 ».
L'Organisation mondiale de la santé en abandonne l'usage en 1965, faisant ainsi suite à une requête informelle de la délégation de la République populaire mongole, pour adopter la dénomination officielle de Down syndrome ou syndrome de Down[55],[65].
Au début des années 1960, les chercheurs découvrent aussi l'existence de formes plus rares de syndrome de Down (trisomie en mosaïque et trisomie non libre par translocation)[52].
Avancées de la fin du XXe siècle
Ces avancées s'effectuent de façon concomitante dans deux domaines : le dépistage prénatal et la génétique moléculaire.
À partir de 1969, l'amniocentèse est utilisée pour l'analyse du caryotype fœtal. Ce type de prélèvement invasif est facilité par le développement de l'échographie obstétricale[62]. Dans les années 1980, des méthodes d'analyses non invasives apparaissent progressivement : marqueurs sériques et signes échographiques qui permettent de suspecter une trisomie 21[68]. À la fin des années 1990, une nouvelle approche diagnostique apparait, basée sur diverses méthodes d'analyse de l'ADN fœtal[22],[62].
En 1993, une étude suggère que les gènes liés aux caractéristiques importantes du syndrome de Down se trouvent dans une région critique du bras long du chromosome 21, définie en 1997 comme DCR-1 (Down Critical Region-1) bientôt subdivisée en sous-régions. Dans le cadre du Projet Génome Humain, le séquençage du chromosome 21 est publié en 2 000, c'est le second chromosome humain décodé après le chromosome 22[69].
Recherches au XXIe siècle
L'utilisation de modèles souris (souris transgéniques) permet de reproduire en grande partie un syndrome de Down chez l'animal. Chez la souris, les gènes homologues de la trisomie 21 humaine sont situés principalement sur son chromosome 16, mais aussi sur les 10 et 17. Plusieurs lignées de souris trisomiques ont été mises au point, une des plus utilisées est celle de la souris Ts65Dn dont on a pu démontrer un déficit important d'apprentissage et de mémoire[19],[69].
Ces modèles souris permettent de « disséquer » les gènes liés à différents phénotypes de trisomie 21, comme l'apparition de maladie d'Alzheimer chez les trisomiques âgés, les malformations cardiaques ou la protection contre des tumeurs solides[19],[69]. 200 à 300 gènes seraient potentiellement responsables de 25 phénotypes de Down.
La trisomie 21 se définit alors comme surexpression de certains gènes, situés sur une région (21q21 à 21q22.3) du bras long du chromosome 21, dont les plus importants contrôlent le développement des structures cérébrales et cardiaques. Ce qui ouvre des perspectives thérapeutiques par recours à des inhibiteurs de la surexpression des gènes en cause[22],[69].
La recherche tente actuellement non pas de supprimer les chromosomes en trop, mais d'annuler leur influence. Plusieurs pistes thérapeutiques sont actuellement à l’étude, dont certaines donnent déjà lieu à des essais cliniques : l’un, mené en Espagne, concerne l’enzyme DYRK1A (Dual specificity tyrosine-phosphorylation-regulated kinase 1A) (en)[70], l’autre, mené par les laboratoires Roche, concerne le neurotransmetteur GABA[71]. Par ailleurs, la Fondation Jérôme-Lejeune a déposé en 2011 un brevet sur une molécule potentiellement thérapeutique[72]. Chez la souris de laboratoire traitée dès la naissance un agoniste de la signalisation hedgehog améliore le comportement et les résultats à l'épreuve du labyrinthe de Morris[73].
Tumeurs
La trisomie 21 diminuerait le risque de développer des tumeurs solides[74]. Cela serait (au moins pour les cancers intestinaux) la sur-expression d’un gène présent en trois exemplaires (du moins dans un modèle expérimental de souris de laboratoire caractérisée par une centaine de gènes orthologues du chromosome humain 21), qui réprime (de 44 % chez cette souris) la croissance tumorale dans les cancers intestinaux. Le gène Ets2, déjà identifié comme oncogène semble en cause (quand il est inactivé chez des souris mutées, le nombre de tumeurs devient inversement proportionnel au nombre de copies de Ets2 fonctionnelles, et à la quantité de ses ARN messagers)[75].
Mémoire
Les personnes porteuses de la trisomie 21 auraient une moindre activité du gyrus denté de l'hippocampe. Ceci serait l'une des explications de la déficience variable de la mémoire de ces sujets. Des gènes sur-exprimés seraient en cause et notamment ceux de la cystathionine bêta-synthase (CBS) et DYRK1A. Des travaux pour mettre au point des inhibiteurs de la CBS et DYRK1A sont en cours.
Par ce souci de mémoire, ils ont besoin de repères pour évoluer plus facilement : ils ont des difficultés à « s’adapter » correctement.
D'autres gènes sont en rapport avec la survenue plus précoce d'une maladie d'Alzheimer. Les travaux en cours sur les bases génétiques et moléculaires de la trisomie 21 sont susceptibles d'être bénéfiques non seulement pour les trisomiques eux-mêmes, mais aussi pour la collectivité tout entière[69].
Développements internationaux
Collaborations scientifiques
Le diagnostic génétique prénatal non invasif et les soins aux trisomiques tendent à s'harmoniser à l'échelle internationale, du moins dans les pays développés. La prise en compte plus précise des variations individuelles de trisomie 21 entraîne des changements de paradigmes, par exemple[76] :
- Les trisomiques 21 ne sont plus considérés comme un groupe homogène indistinct, chaque trisomique doit bénéficier d'une approche individuelle personnalisée lui permettant d'exprimer le maximum de ses capacités. La plupart des problèmes médicaux peuvent être traités efficacement.
- Cette approche n'est plus seulement biomédicale, ou de génétique médicale, mais multidisciplinaire car le développement neurocognitif dépend beaucoup de l'environnement familial et du contexte psychosocial.
Ces nouvelles approches nécessitent de vastes essais cliniques pour être validées, d'où une collaboration internationale pour harmoniser et mettre en commun les moyens et les méthodes. En Europe et aux États-Unis, quinze sites conduisent des essais cliniques, coordonnés par des plateformes telles que Horizon21 DS Consortium[77], d'autres initiatives se font sous l'égide d'organismes privés et publics collaborant avec des universités comme avec l'industrie pharmaceutique. En 2019, le NIH américain alloue un budget de 70 millions $ (contre 29 millions $ en 2001) à la recherche sur le syndrome de Down et ses comorbidités[52].
Institutions et société civile
En , l'Assemblée générale des Nations unies a décidé de proclamer le Journée mondiale de la trisomie 21, qui sera célébrée chaque année à partir de 2012. Cette date du est hautement symbolique : elle fait référence aux 3 chromosomes 21 à l'origine de la maladie. Le but de cette Journée est d'inviter tous les États Membres, les organismes compétents du système des Nations-Unies et les autres organisations internationales, ainsi que la société civile, y compris les organisations non gouvernementales et le secteur privé, à sensibiliser et informer l'opinion publique sur la trisomie 21.
Une campagne européenne, « Chère Future Maman » (Dear Future Mum), financée et soutenue par une dizaine d’associations dont la Fondation Jérôme-Lejeune, controversée pour ses positions anti-avortement, est aussi lancée le , appuyée sur un court-métrage qui rassemble des acteurs italiens, espagnols, anglais et français tous porteurs de trisomie 21. Mise en ligne le sur le site de partage YouTube, cette vidéo a été visionnée plus de quatre millions de fois en moins d'une semaine. En France, le CSA décide de suspendre la diffusion de cette vidéo car susceptible de troubler les femmes ayant eu recours à une interruption médicale de grossesse.
Aspects socioculturels
Les conséquences de la trisomie 21 dépendent beaucoup de facteurs individuels et familiaux, comme pour tous les enfants, mais aussi de la qualité de la pertinence des rééducations précoces ou pas, des accompagnements spécifiques et du regard porté sur la personne porteuse de trisomie 21. Par ailleurs, lorsque l'adulte éducateur pense que l'enfant n'arrivera pas à un niveau d'autonomie sur un point précis, la probabilité est plus forte que l'enfant en effet n'y arrive pas (cet effet est décrit en psychologie comme l'effet pygmalion). Ce facteur accentue fortement les disparités entre les personnes trisomiques, les adultes éducateurs, parents en général, ayant des difficultés à imaginer jusqu'où leur enfant peut aller.
D'une manière générale, on peut noter que la trisomie 21 entraîne un développement neuronal différent, avec une quantité de connexions plus faibles que pour les personnes non porteuses[78]. Ceci entraîne une capacité de traitement des informations plus faible, et donc un délai de traitement plus long. On observe donc très souvent chez la personne porteuse de trisomie 21 un temps de latence, plus ou moins important, entre l'information reçue et la réaction à celle-ci. Ceci explique aussi en partie les difficultés d'expression rencontrées par les personnes porteuses de trisomie 21, qui mettent souvent un peu de temps à exprimer de manière claire ce qu'elles veulent dire. Cet état de fait peut amener certains à écrire ceci : « Entre autres règles de communication, il est recommandé de s'exprimer par des phrases claires et concises, de répondre aux questions avec pertinence et dans un délai court. »
Les personnes atteintes de trisomie 21 doivent surmonter les conséquences de leur retard physique et intellectuel, mais plus souvent encore le regard inquiet ou hostile que certains portent sur leur anomalie qui n'est pourtant pas contagieuse et qui est plus familière et moins désespérante pour les familles que bien des affections congénitales.
Il est nécessaire pour les familles de solliciter le conseil de spécialistes qui les aideront à susciter et à encourager les progrès intellectuels et physiques (rééducation musculaire, activités physiques adaptées) du sujet. Il y a autant d'approches que de pays, et en France même plusieurs pratiques se mélangent. En conséquence, les tentatives de généralisation peuvent porter à polémique. Il est généralement admis que des rééducations précoces en orthophonie et kinésithérapie sont utiles. Au Brésil, il est préconisé jusqu'à 4 heures de kinésithérapie par semaine pour le bébé porteur de trisomie 21, de façon qu'il puisse aller à l'école au même âge que les autres et avec les mêmes capacités motrices. Cette approche est vue comme très contraignante pour l'enfant en France.
Les enfants atteints de la trisomie 21 ont été longtemps cachés. Leur existence était alors malheureuse et très brève, leur espérance de vie moyenne ne dépassait pas l'adolescence. Autant que possible il est conseillé de ne pas cantonner les enfants trisomiques à la fréquentation d'un milieu spécialisé : de nombreuses études montrent que le contact de l'école et d'enfants sains a une influence extrêmement positive sur le développement général et sur le quotient intellectuel du sujet atteint de trisomie (QI allant de 20 à 80 environ, selon divers paramètres, notamment les stimulations intellectuelles offertes par l'environnement). Avec une prise en compte de leurs insuffisances physiologiques et en les traitant comme les êtres pensants et aimants qu'ils sont, la médecine parvient aujourd'hui à leur donner une espérance de vie presque normale et il n'est pas rare qu'ils prennent une place dans la vie active.
Certains trisomiques sont capables de s'intégrer à la société de façon autonome à l'âge adulte. Certains ont un QI dit « normal » (supérieur à 70).
Une approche pédagogique et psychothérapeutique inspirée de la psychanalyse et visant à réparer les blessures narcissiques dont souffrent les trisomiques, tout en leur permettant des progrès cognitifs, est développée par une psychanalyste du GIREP, Madeleine Natanson qui en fait le sujet de sa thèse en 1987 : « Réparation symbolique et alliance pédagogique[79] ».
Représentations dans l'art

- Le peintre Andrea Mantegna (1431-1506) a eu quatorze enfants, dont un aurait été atteint de trisomie 21, et qui pourrait figurer dans plusieurs de ses œuvres, par exemple dans La Vierge à l'Enfant entre saint Jérôme et saint Louis de Toulouse[80].
- Dans un tableau peint en 1515, L’Adoration de l’enfant Jésus, un peintre anonyme, élève du maître flamand Jan Joest van Kalkar, représente un ange ayant des traits physiques évoquant une trisomie 21[80].
- Dans un tableau de Jacob Jordaens (1593-1678), le mangeur de porridge (vers 1660)[80],[81].
- En 1998, le photographe russe Rauf Mamedov (az) (né en 1956 en Azerbaïdjan) revisite la Cène peinte par Léonard de Vinci. Les personnages existants sont remplacés par des trisomiques.
Représentation au cinéma ou à la télévision
- Corky, un adolescent pas comme les autres (1989-1993), série télévisée américaine créée par Michael Braverman
- L'Hôpital et ses fantômes (1994 - 1997) , série danoise réalisée par Lars Von Trier
- Le Huitième Jour (1996), film belge réalisé par Jaco Van Dormael
- Florian - Un enfant à aimer (1999), téléfilm allemand réalisé par Dominique Othenin-Girard
- Des tas de choses (2004), court-métrage documentaire suisse réalisé par Germinal Roaux
- Yo, también (2010), film espagnol réalisé par Álvaro Pastor et Antonio Naharro
- Café de Flore (2012), film franco-québécois réalisé par Jean-Marc Vallée
- Sweetheart (Deux sœurs amoureuses) (2012), court-métrage britannique réalisé par Eva Riley
- El carro azul (de) (La Voiture bleue) (2014), court-métrage germano-cubain réalisé par Valerie Heine
- L'École de la vie (2016), documentaire chilien réalisé par Maite Alberdi
- Mention particulière (2017), téléfilm français réalisé par Christophe Campos
- Champions (Campeones) (2018), film espagnol réalisé par Javier Fesser
- Glee (2009), série télévisée américaine créée par Fox Broadcasting Company
- The Peanut Butter Falcon (2020)
- Mes premières fois (2020), série télévisée américaine, avec Lily D. Moore dans le rôle de Rebecca
Représentation en littérature
- Ce n'est pas toi que j'attendais (2014) roman graphique autobiographique de Fabien Toulmé
Cas notoires

Politiques et apparentés
- Anne de Gaulle[82].
- Pablo Pineda, première personne trisomique à obtenir une licence en psycho-pédagogie, conseiller social à la mairie de Malaga et enseignant, élu « personnalité de l'année » 2004 par le journal El País, Coquille d'argent du meilleur acteur du Festival international du film de Saint-Sébastien pour son rôle dans le film Yo, también (2009). Il définit la trisomie comme « une caractéristique personnelle supplémentaire ».
- Angela Bachiller (en), conseillère municipale à la municipalité de Valladolid, Espagne ()[83],[84].
- Éléonore Laloux : porte-parole du Collectif les Amis d'Éléonore. Elle est intervenue dans plusieurs débats publics sur le thème de la trisomie 21. En , elle publie un livre-témoignage aux Max Milo Éditions, Triso et alors !, en collaboration avec Yann Barte.
Artistes
- Chris Burke, acteur américain connu pour avoir joué pendant quatre saisons le rôle de Corky dans la série télévisée Corky, un adolescent pas comme les autres (1989).
- Pascal Duquenne, acteur, lauréat (ex-æquo avec Daniel Auteuil) du prix d'interprétation masculine au Festival de Cannes 1996 pour son interprétation dans le film Le Huitième Jour de Jaco Van Dormael. En 2011, les éditions Pictura ont publié Monotypes, un livre d'art consacré aux gravures de Pascal Duquenne[85].
- Nathalie Nechtschein, écrivaine et poétesse française, dont l'un des textes se retrouve sur l'album D'Elles de Céline Dion.
- Luke Zimmerman (en), acteur américain qui joue dans la série La Vie secrète d'une ado ordinaire qui a débuté en 2008 et qui est arrivée maintenant à la quatrième saison.
- Stephane Ginnsz, un des premiers acteurs trisomiques à obtenir le premier rôle dans un film, intitulé Duo: The True Story of a Gifted Child with Down Syndrome, sorti en 1996, racontant l'histoire d'un jeune garçon trisomique amoureux d'une violoniste et dont le rêve est de pouvoir un jour l'accompagner au piano. Ce film est recompensé la même année du prix Martin-Scorsese et du prix Warner Bros Pictures, finaliste au Festival international de films de Chicago pour les enfants et nominé pour un prix de l'Association pour personnes à handicaps sévères (en) (TASH Award) (1997).
- Jamie Brewer, actrice américaine née en 1985[86], connue pour ses rôles dans American Horror Story. En , Jamie Brewer défilait pour la créatrice Carrie Hammer sur un podium de Fashion Week.
- Lauren Potter, actrice américaine née en 1990[87]. Elle a notamment joué le rôle de Becky Jackson dans la série télévisée Glee.
- Maryam Alakbarli, artiste-peintre originaire d'Azerbaïdjan. Elle a exposé à Rome, Paris, Moscou et Deauville[88]. En 2011, elle a été admise à l'École nationale supérieure des arts décoratifs de Paris.
- Marin Gerrier, acteur connu pour avoir joué dans le film Franco québécois le Café de Flore de Jean-Marc Vallée.
Notes et références
- ↑ (en) MMWR Morb Mortal Wkly Rep. 1994, Olsen CL, Cross PK, Gensburg LJ, Hughes JP., 1996.
- ↑ « Trisomie 21 - Génétique », sur www.msss.gouv.qc.ca Santé et services sociaux Québec, (consulté le ).
- ↑ (en) Herman et al. « Beside estimation of Down syndrome risk during first trimester ultrasound screening » Ultrasound Obstet Gynecol. 2002; 20:468-475. Résumé, Texte complet[PDF].
- ↑ (en) Kallen B, Mastroiacovo P et Robert E., 1996.
- ↑ (en) Diamond LS, Lynne D et Sigman B, 1981.
- ↑ (en) Ferguson-Smith M.A « Prenatal chromosome analysis and its impact on the birth incidence of chromosome disorders » Br Med J. 1983;4:355-64.
- ↑ « Trisomie 21 : le grand remue-méninges » dans Le Journal du CNRS.
- 1 2 3 4 5 (en) Jensen KM, Bulova PD, Managing the care of adults with Down’s syndrome, BMJ, 2014;349:g5596.
- ↑ (en) Natoli JL, Ackerman DL, McDermott S, Edwards JG, Prenatal diagnosis of Down syndrome: a systematic review of termination rates (1995-2011), Prenat Diagn, 2012;32:142-53.
- ↑ Natural history of Down’s syndrome: a brief review for those involved in antenatal screening Joan Noble 1998 DOI 10.1136/jms.5.4.172
- ↑ Dr Bénédicte De Freminville, Trisommie 21 : Résumé sur le site orphanet, 2007 (lire en ligne).
- ↑ (en) Malt, EA; Dahl, RC; Haugsand, TM; Ulvestad, IH; Emilsen, NM; Hansen, B; Cardenas, YE; Skøld, RO; Thorsen, AT; Davidsen, EM (5 février 2013). "Health and disease in adults with Down syndrome". Tidsskrift for den Norske laegeforening : tidsskrift for praktisk medicin, ny raekke 133 (3): 290–4. doi:10.4045/tidsskr.12.0390. .
- 1 2 3 4 5 6 7 8 9 10 11 12 13 Faisal Akhtar et Syed Rizwan A. Bokhari, « Down Syndrome », dans StatPearls, StatPearls Publishing, (PMID 30252272, lire en ligne)
- ↑ Bénédicte De Freminville et Renaud Touraine, Trisomie 21, 2007, sur le site orpha.net.
- 1 2 3 4 5 6 7 8 9 10 11 12 13 (en) Marilyn J. Bull, « Down Syndrome », The New England Journal of Medicine, vol. 382, no 24, , p. 2344–2352 (ISSN 1533-4406, PMID 32521135, DOI 10.1056/NEJMra1706537)
- 1 2 3 4 5 6 7 8 9 10 11 Renaud Touraine, « A propos d'une maladie chromosomique : la trisomie 21 », La Revue du Praticien, vol. 60, , p. 709-714.
- ↑ Dimopoulos K, Constantine A, Clift P et al. Cardiovascular complications of Down syndrome: Scoping review and expert consensus, Circulation, 2023;147:425–441
- ↑ (en) Steingass KJ, Chicoine B, McGuire D, Roizen NJ, Developmental disabilities grown up: Down syndrome, J Dev Behav Pediatr, 2011;32:548-58.
- 1 2 3 4 5 6 7 8 9 10 Stylianos E. Antonarakis, Brian G. Skotko, Michael S. Rafii et Andre Strydom, « Down syndrome », Nature Reviews. Disease Primers, vol. 6, no 1, , p. 9 (ISSN 2056-676X, PMID 32029743, PMCID 8428796, DOI 10.1038/s41572-019-0143-7, lire en ligne, consulté le )
- ↑ Geijer JR, Stanish HI, Draheim CC, Dengel DR, Bone mineral density in adults with Down syndrome, intellectual disability, and nondisabled adults, Am J Intellect Develop Disabil, 2014;119:107-14.
- 1 2 3 Aimé Ravel, « Trisomie 21 : surveillance par le généraliste : de l'adolescence à la fin de la vie », Le Concours Médical, vol. 126, no 21, , p. 1196-1198.
- 1 2 3 Ambreen Asim, Ashok Kumar, Srinivasan Muthuswamy et Shalu Jain, « “Down syndrome: an insight of the disease” », Journal of Biomedical Science, vol. 22, no 1, , p. 41 (ISSN 1021-7770, PMID 26062604, PMCID 4464633, DOI 10.1186/s12929-015-0138-y, lire en ligne, consulté le )
- ↑ Aimé Ravel, « Trisomie 21 : surveillance par le généraliste : de 1 an à l'adolescence », Le Concours Médical, vol. 126, no 19, , p. 1086-1088.
- ↑ « Cette thérapie représente un réel espoir pour les personnes porteuses de trisomie 21 », sur Le HuffPost, (consulté le )
- 1 2 3 4 5 Alain Verloes, « A propos d'une maladie chromosomique : la trisomie 21 », La Revue du Praticien, vol. 54, no 12, , p. 1363-1370.
- ↑ P. Winders, K. Wolter-Warmerdam et F. Hickey, « A schedule of gross motor development for children with Down syndrome », Journal of intellectual disability research: JIDR, vol. 63, no 4, , p. 346–356 (ISSN 1365-2788, PMID 30575169, DOI 10.1111/jir.12580, lire en ligne, consulté le )
- ↑ Karin Windsperger et Stefanie Hoehl, « Development of Down Syndrome Research Over the Last Decades–What Healthcare and Education Professionals Need to Know », Frontiers in Psychiatry, vol. 12, (ISSN 1664-0640, PMID 34970162, PMCID PMC8712441, DOI 10.3389/fpsyt.2021.749046, lire en ligne, consulté le )
- ↑ (en) Jin Liang Zhu, Carsten Obel, Henrik Hasle et Sonja A. Rasmussen, « Social conditions for people with Down syndrome: A register-based cohort study in Denmark », American Journal of Medical Genetics Part A, vol. 164, no 1, , p. 36–41 (PMID 24273114, PMCID PMC4490827, DOI 10.1002/ajmg.a.36272, lire en ligne, consulté le )
- ↑ Lire en ligne article gratuit.
- ↑ Le test génétique non invasif de la trisomie 13, 18 et 21 fœtale sur le site expertadn.fr lire en ligne.
- ↑ Voir article sur medscape.fr lire en ligne.
- ↑ (en) Caroline Mansfield et al. « Termination rates after prenatal diagnosis of Down syndrome, spina bifida, anencephaly, and Turner and Klinefelter syndromes: a systematic literature review » Prenatal Diagnosis 1999 (vol. 19 no 9, p. 808–812 résumé en ligne.
- ↑ (en) David W. Britt, et al. « Determinants of parental decisions after the prenatal diagnosis of Down syndrome: Bringing in context » American Journal of Medical Genetics 1999 (vol. 93 no 5, p. 410–416), résumé en ligne.
- ↑ Le Quotidien du médecin (Antoine Dalat) 12/05/2011.
- ↑ Laura Hercher, « Des riches génétiquement modifiés », sur Le Monde diplomatique,
- ↑ (en) James P Neilson et Zarko Alfirevic « Optimising prenatal diagnosis of Down's syndrome » BMJ 2006;332:433-4 (25 February) DOI 10.1136/bmj.332.7539.433.
- ↑ « Pourquoi dépister autrement les trisomies », sur sante.lefigaro.fr (consulté le ).
- ↑ .
- 1 2 Catherine Bachelard-Jobard, L'eugénisme, la science et le droit, Presses Universitaires de France, coll. « Partage du savoir », , 368 p. (ISBN 978-2-13-063850-6 et 2-13-063850-3, OCLC 907647395, lire en ligne), p. 117-133.
- ↑ « Le diagnostic prénatal engendre-t-il une nouvelle forme d'eugénisme ? », sur France Culture (consulté le ).
- ↑ Jacques Milliez, L'euthanasie du foetus : médecine ou eugénisme ?, Paris, Odile Jacob, coll. « Médecine », , 218 p. (ISBN 2-7381-0682-X et 9782738106827, lire en ligne), p. 62 ; 153.
- ↑ La trisomie est une tragédie grecque, éditions Salvator, 2009.
- ↑ , Éloge des phénomènes, éditions Max Milo, 2014.
- ↑ .
- ↑ (en) Diamandopoulos A.A. et al. « A Neolithic case of Down syndrome » Journal of the History of Neuroscience 1997;6(1):86-9.
- 1 2 Philippe Charlier, Médecin des morts : récits de paléopathologie, Paris, Fayard, , 394 p. (ISBN 978-2-213-62722-9), chap. 7, p. 93-100.
- ↑ (en) Czarnetzki A, Blin N, Putsch CM. « Down's syndrome in ancient Europe » The Lancet 2003;362(9388):1000. .
- ↑ P. Barbet, J. L. Fisher et C. Jacques, « Les Nains d'Angkor » Pour la Science, août 2007, p. 96-97.
- ↑ F. Dunand et R. Lichtenberg, Momies d'Égypte et d'ailleurs, Paris, Éditions du Rocher Champollion, .
- ↑ Des maladies mentales considérées sous le rapport médical, hygiénique et médico-légal", Paris, Jean-Baptiste Baillière, 1838, 2 volumes.
- ↑ Traitement moral, hygiène et éducation des idiots et des autres enfants arriérés ou retardés dans leur développement, Jean-Baptiste Baillière, Paris, 1846.
- 1 2 3 4 Melissa J. Alldred, Alessandra C. Martini, David Patterson et James Hendrix, « Aging with Down Syndrome—Where Are We Now and Where Are We Going? », Journal of Clinical Medicine, vol. 10, no 20, , p. 4687 (ISSN 2077-0383, PMID 34682809, PMCID 8539670, DOI 10.3390/jcm10204687, lire en ligne, consulté le )
- ↑ (en) Down, J.L.H., « Observations on an ethnic classification of idiots », Clinical Lecture Reports, London Hospital, vol. 3, , p. 259–262 (résumé)
- 1 2 3 4 5 (en) K.F. Kiple (dir.) et Christine E. Cronk, The Cambridge World History of Human Disease, Cambridge, Cambridge University Press, , 1176 p. (ISBN 0-521-33286-9), partie VIII, chap. 37 (« Down Syndrome »), p. 683-686.
- 1 2 3 (en) O. Ward, « John Langdon Down: The Man and the Message », Down Syndrome Research and Practice, vol. 6, no 1, , p. 19–24 (ISSN 0968-7912, DOI 10.3104/perspectives.94, lire en ligne, consulté le )
- ↑ (en) Stefan Kutzsche, « John Langdon Down (1828-1896) - a pioneer in caring for mentally disabled patients », Acta Paediatrica, vol. 107, no 11, , p. 1851–1854 (DOI 10.1111/apa.14505, lire en ligne, consulté le )
- ↑ (en-US) « Normansfield – Langdon Down Museum » (consulté le )
- ↑ Éloge des phénomènes, Bruno Deniel-Laurent, Paris, Max Milo, 2014 (pages 15-16).
- ↑ (en) « Kalmuc idiocy : report of a case with autopsy / by John Fraser ; with notes on sixty-two cases by Arthur Mitchell. », sur Wellcome Collection (consulté le )
- 1 2 3 4 5 6 7 Marie-Hélène Couturier-Turpin, « La découverte de la trisomie 21 », La Revue du Praticien, vol. 55, no 12, , p. 1385-1389.
- ↑ K. Codell Carter, « Early conjectures that Down syndrome is caused by chromosomal nondisjunction », Bulletin of the History of Medicine, vol. 76, no 3, , p. 528–563 (ISSN 0007-5140, PMID 12486916, DOI 10.1353/bhm.2002.0118, lire en ligne, consulté le )
- 1 2 3 (en) Malcolm A. Ferguson-Smith, « Cytogenetics and the evolution of medical genetics », Genetics in Medicine, vol. 10, no 8, , p. 553–559 (ISSN 1098-3600 et 1530-0366, PMID 18641515, DOI 10.1097/GIM.0b013e3181804bb2, lire en ligne, consulté le )
- ↑ Lejeune J, Gautier M, Turpin R, « Étude des chromosomes somatiques de neuf enfants mongoliens », Comptes Rendus Hebd Seances Acad Sci, vol. 248, no 11, , p. 1721–1722 (résumé)
- ↑ Wright, David, 1965-, Down's syndrome : the history of a disability, Oxford University Press, (ISBN 978-0-19-157327-9 et 0191573272, OCLC 752614754, lire en ligne)
- 1 2 N Howard-Jones, « On the diagnostic term "Down's disease". », Medical History, vol. 23, no 1, , p. 102–104 (ISSN 0025-7273, PMID 153994, PMCID 1082401, lire en ligne, consulté le )
- ↑ (en) ML Rodríguez-Hernández et Montoya, E, « Fifty years of evolution of the term Down's syndrome », Lancet, vol. 378, no 9789, , p. 402 (PMID 21803206, DOI 10.1016/s0140-6736(11)61212-9).
- ↑ Gordon Allen, C. E. Benda, J. A. Böök et C. O. Carter, « Mongolism », American Journal of Human Genetics, vol. 13, no 4, , p. 426 (ISSN 0002-9297, PMID 17948460, PMCID 1932135, lire en ligne, consulté le )
- ↑ J.M. Thoulon, « Les signes d'appel de la trisomie 21 : Vers un dépistage pendant la vie foetale », La Revue du Praticien, vol. 41, no 21, , p. 2062-2064.
- 1 2 3 4 5 (en) André Mégarbané, Aimé Ravel, Clotilde Mircher et Franck Sturtz, « The 50th anniversary of the discovery of trisomy 21: The past, present, and future of research and treatment of Down syndrome », Genetics in Medicine, vol. 11, no 9, , p. 611–616 (ISSN 1098-3600 et 1530-0366, DOI 10.1097/GIM.0b013e3181b2e34c, lire en ligne, consulté le )
- ↑ (en) « Egcg, a dyrk1a Inhibitor as Therapeutic Tool for Reversing Cognitive Deficits in Down Syndrome Individuals », sur clinicaltrials.gov (consulté le ).
- ↑ (en) Martínez-Cué C, Martínez P, Rueda N, Vidal R, García S, Vidal V, Corrales A, Montero JA, Pazos Á, Flórez J, Gasser R, Thomas AW, Honer M, Knoflach F, Trejo JL, Wettstein JG, Hernández MC., « Reducing GABAA a5 receptor-mediated inhibition rescues functional and neuromorphological deficits in a mouse model of down syndrome », J Neurosci., vol. 33, no 9, , p. 3953-3966 (PMID 23447605, DOI 10.1523/JNEUROSCI.1203-12.2013, lire en ligne).
- ↑ Les différents programmes de recherche de la Fondation Lejeune.
- ↑ (en) Das I, Park LM, Shin JH, Jeon SK, Lorenzi H, Linden DJ, Worley PF, Reeves RH., « Hedgehog Agonist Therapy Corrects Structural and Cognitive Deficits in a Down Syndrome Mouse Model », Sci Transl Med., vol. 5, no 201, , p. 201ra120 (DOI 10.1126/scitranslmed.3005983).
- ↑ (en) Yang Q. et al. « Mortality associated with Down's syndrome in the USA from 1983 to 1997: a population-based study » Lancet 2002;359(9311):1019-25 .
- ↑ (en) Sussan TE, Yang A, Fu Li, Ostrowski MC, Reeves RH, Trisomy represses ApcMin-mediated tumours in mouse models of Down's syndrome, Nature, 2008;451:73-5.
- ↑ Karin Windsperger et Stefanie Hoehl, « Development of Down Syndrome Research Over the Last Decades-What Healthcare and Education Professionals Need to Know », Frontiers in Psychiatry, vol. 12, , p. 749046 (ISSN 1664-0640, PMID 34970162, PMCID 8712441, DOI 10.3389/fpsyt.2021.749046, lire en ligne, consulté le )
- ↑ « Home », sur Horizon 21 (consulté le )
- ↑ (en) Weick JP, Held DL, Bonadurer GF 3rd, Doers ME, Liu Y, Maguire C, Clark A, Knackert JA, Molinarolo K, Musser M, Yao L, Yin Y, Lu J, Zhang X, Zhang SC, Bhattacharyya A., « Deficits in human trisomy 21 iPSCs and neurons », Proc Natl Acad Sci U S A., vol. 110, no 24, , p. 9962-9967 (PMID 23716668, DOI 10.1073/pnas.1216575110, lire en ligne).
- ↑ Madeleine Natanson, Réparation symbolique et alliance pédagogique : Réflexion à partir de l'écoute de jeunes trisomiques, Fleurus, 1988. (ISBN 2-215-01228-5).
- 1 2 3 André Stahl et Pierre Tourame, « L'ancienneté de la trisomie 21 et sa représentation dans les arts visuels », Histoire des sciences médicales, vol. 47, no 1, , p. 19-28 (lire en ligne)
- ↑ La source Stahl indique, à tort semble-t-il, qu'il s'agit du tableau Le Satyre et les Paysans. On peut comparer les deux tableaux sur Wikimedia Commons
- ↑ (en) Jonathan Fenby « The little girl who conquered de Gaulle » sur http://www.thetimes.co.uk, .
- ↑ (es) Ángela Bachiller es la primera edil con síndrome de Down, sur http://www.abc.es, article du .
- ↑ (es) « Ángela Bachiller se convierte en la primera concejal con síndrome de Down »(Archive.org • Wikiwix • Archive.is • Google • Que faire ?) (consulté le ) Sur sociedad.elpais.com, article du .
- ↑ [PDF] Présentation du livre Monotypes de Pascal Duquenne.
- ↑ (en) Actors with Down Syndrome Raise Awareness, ABCnews, .
- ↑ (en) Glee' star tapped to join Obama's committee, CNN, .
- ↑ , Vivre FM, .
Annexes
Bibliographie
- Bruno Deniel-Laurent, Éloge des phénomènes, Max Milo Éditions, Paris, 2014.
- Éléonore Laloux et Yann Barte, Triso et alors !, Max Milo Éditions, Paris, 2014.
- Jean-Jacques d’Amore et Delphine Vasseur, Supplément d’âme – Trisomie 21 le chromosome en +, Éditions Degeorge, Arras, 2011 ; Max Milo éditions, 2020 (ISBN 2315005035)
- Monique Cuilleret, Trisomie et handicaps génétiques associés, Éditions Masson, 2006 (ISBN 2294755987)
- Pr. Marie-Odile Réthoré, Dr. Henri Bléhaut, Dr. Sylvie de Kermadec, et alii, Trisomie 21, guide à l’usage des familles et de leur entourage, Éditions Bash, janvier 2006 (ISBN 2845040415)
- Trisomie 21 France, 100 idées pour en savoir plus sur les personnes avec trisomie 21 … et casser les idées reçues, Tom Pousse, 2017, (ISBN 2353451802)
Articles connexes
Liens externes
- Fiche résumé de la Trisomie 21 sur Orphanet
- Trisomie 21, guide sur le site de la Haute Autorité de santé