
L'amplification en chaîne par polymérase (ACP)[1] ou réaction de polymérisation en chaîne[2], généralement siglée PCR (de l'anglais : polymerase chain reaction) est une méthode de biologie moléculaire permettant d'obtenir rapidement, in vitro, un grand nombre de segments d'ADN identiques, à partir d'une séquence initiale[3],[4]. Le terme test d'amplification des acides nucléiques (TAAN), très peu utilisé par les chercheurs (moins de 200 articles référencés dans Google Scholar en , contre plus de 45 000 pour la requête PCR dans les pages en français), est plus restrictif, car il désigne l'application de la PCR à des fins diagnostiques. Or, cette technique a de multiples applications en recherche, dès lors qu'il est nécessaire d'obtenir des quantités notables d'ADN à partir d'une source limitée. Il est aussi quelque peu incorrect, car il serait préférable d'écrire test par amplification des acides nucléiques : en effet, ce n'est pas l'amplification qui est testée, elle est le moyen permettant le test.
Elle permet de multiplier en grand nombre (avec un facteur de l'ordre du milliard) une séquence d'ADN ou d'ARN connue (avec une étape supplémentaire de rétrotranscription pour l'ARN), à partir d'une faible quantité (de l'ordre de quelques picogrammes) d'acide nucléique, et à l'aide d'amorces spécifiques constituées d'oligonucléotides de synthèse de dix-sept à vingt-cinq nucléotides. Le terme amplicon désigne aussi bien la séquence de départ que le produit obtenu. Cette technique permet, par exemple, de détecter la présence du VIH ou mesurer une charge virale (concentration du virus dans le plasma), des traces d'OGM (organismes génétiquement modifiés), ou encore des virus d'hépatites B, C et D. Elle permet également de préparer des quantités notables d'un fragment donné à des fins d'insertion dans des vecteurs ad hoc (clonage).
De plus en plus utilisée en criminalistique, cette technique se fonde sur la combinaison de deux facteurs :
- les propriétés de synthèse enzymatique et d’initiation spécifique à l'ADN double brin spécifique des ADN polymérases dépendantes à l'ADN thermostables ;
- les propriétés d’hybridation et de déshybridation des brins complémentaires d’ADN en fonction de la température.
Ces éléments permettent de contrôler l’activité enzymatique grâce à des transitions de température (assurées par un thermocycleur) répétées de manière cyclique (cf. réaction en chaîne).
Les premières ADN polymérases utilisées en PCR provenaient d'une bactérie thermophile (résistante à des températures très élevées), par exemple Thermus aquaticus (Taq polymérase) ou encore Pyrococcus furiosus (Pfu polymérase), Thermococcus litoralis (Vent ou Tli polymérase), Thermus thermophilus (Tth polymérase). De nos jours, les enzymes utilisées sont dites recombinantes, ce qui simplifie considérablement leur obtention, et leurs propriétés ont été largement modifiées pour les rendre plus efficaces, plus fidèles.
En moins de dix ans, on a appris à faire plus d'un milliard de copies en moins d'une heure, ce qui a imposé la PCR dans les laboratoires en révolutionnant la biologie moléculaire. Elle n'est cependant pas fiable pour certaines sources d'échantillons (urine[5], crachats[5]), peut-être en raison d'inhibiteurs de la Taq polymérase.
Historique
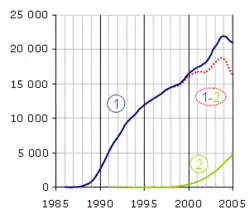
La PCR est une technologie qui a bouleversé la biologie moléculaire et s'est implantée très rapidement dans les laboratoires. En revanche, la PCR en temps réel a dû attendre la mise sur le marché d'un certain nombre d'innovations technologiques avant de se développer et est encore considérée comme une méthodologie nouvelle. Le nombre d'articles par an répondant aux mots clés « polymerase chain reaction » (1) et « real-time polymerase chain reaction » (2) sur le moteur de recherche PubMed donne une assez bonne idée de leur importance dans le monde scientifique. Notez que la méthode n'est pas exempte de biais, par exemple quelques articles sont trouvés pour la PCR en temps réel en 1991 et 1992 (en pointillé) alors que son principe n'a été décrit qu'en 1993. La différence (1-2) est représentative du poids de la PCR en point final, qui va probablement céder progressivement la place au temps réel.
Cette technique a largement évolué depuis ses débuts. Parmi les évolutions les plus fondamentales, on retrouve :
- le remplacement du fragment de Klenow d’ADN polymérase I d’E . coli par une polymérase thermorésistante (initialement la Taq) qui évite de devoir remettre de l’enzyme à chaque cycle. Cette innovation permet un bond énorme vers l’automatisation et évite de devoir ouvrir le tube réactionnel, limitant considérablement le risque de contamination ;
- la généralisation des thermocycleurs (un bon nombre d’anciennes expérimentations ont été réalisées avec trois bains-marie) a permis de rendre la PCR moins contraignante, plus reproductible et était un prérequis indispensable à la plupart des applications nouvelles ;
- l'invention de la PCR en temps réel qui permet de rendre la méthode quantitative et évite plusieurs étapes expérimentales contraignantes, telles l’électrophorèse sur gel d’agarose, l’acquisition de fluorescence, la calibration de l’acquisition du signal, etc.
Par souci de clarté, les dates correspondent à la première publication sur le domaine et seuls les premiers auteurs sont cités, les références complètes étant dans le chapitre « Bibliographie ». Ce choix permet en outre de limiter les polémiques telles le rôle de Rosalind Elsie Franklin dans la découverte de la structure de la double hélice d’ADN.
- 1953 : Découverte de la structure en double hélice de l’ADN par James Dewey Watson et Francis Harry Compton Crick, (prix Nobel de physiologie ou médecine en 1962) d'après les travaux de Rosalind Franklin.
- 1956 : Découverte de l’ADN polymérase ADN dépendante (ADN pol I) par Arthur Kornberg (prix Nobel de physiologie ou médecine en 1959).
- 1970 : Co-découverte indépendante de l’ADN polymérase ARN dépendante par Howard Martin Temin et David Baltimore (prix Nobel de physiologie ou médecine en 1975).
- 1986 : Première publication publique sur la PCR par Kary Mullis (prix Nobel de chimie en 1993).
- 1988 : Première PCR réalisée avec une ADN polymérase thermostable, provenant de Thermus aquaticus, par Saiki RK.
- 1991 : Première détection du produit de PCR par sonde (sonde d’hydrolyse) par Holland PM.
- 1992 : Invention de la PCR en temps réel par Russell Higuchi.
- 1995 : Première publication sur la TAIL-PCR par Liu YG
- 1996 : Mise au point des polymérases temporairement inactives et activables par la chaleur par Birch DE.
- 1997 : Mise en évidence de la variation de température "Tm dépendante" du SYBR green par Wittwer CT.
- 1997 : Première discrimination d’allèle grâce à la courbe de fusion par Lay MJ.
Principe
La PCR est une technique basée sur une répétition de cycles de transition de température. Sauf pour certaines méthodes (par exemple l’utilisation de sondes d’hydrolyse), chaque cycle contient trois étapes détaillées ci-dessous. Par souci de didactisme, nous allons considérer pour l’exemple qui suit une efficacité de PCR de 100 %.

Étapes de la PCR
Conditions natives (0 sur le schéma)
Cette étape se fait généralement à température ambiante. L’ADN bicaténaire adopte sa conformation en double hélice. Dans cet exemple, nous considérerons qu’il n’y a qu’une molécule initiale d’ADN double brin dans la solution, la zone colorée (magenta et orange) correspondant à notre amplicon. Note: les ADN complémentaires issus de la transcription inverse sont généralement simple brin et adopteront alors de complexes conformations tridimensionnelles similaires à celle des ARN.
Dénaturation initiale (1’ sur le schéma)
Avant de commencer les cycles de PCR proprement dit, une étape de chauffage (généralement 10 à 15 minutes à 95 °C) est réalisée. Cette étape permet de : déshybrider les ADN double brin, de casser les structures secondaires, d’homogénéiser le milieu réactionnel par agitation thermique, d’activer les polymérases de type « Hot start », de dénaturer d’autres enzymes qui pourraient être dans la solution (Transcriptase Inverse, Uracil-N-Glycosylase).
Phase de dénaturation (1 sur le schéma)
Cette étape (généralement 0 à 1 minute à 95 °C) permet de déshybrider les ADN, de « décrocher » les polymérases qui seraient encore liées à une matrice et d’homogénéiser le milieu réactionnel.
Phase d’hybridation ou d'appariement des amorces (2 sur le schéma)
Cette étape (généralement 2 à 60 secondes à 56–64 °C) permet aux amorces sens et anti-sens de s’hybrider aux ADN matrice grâce à une température qui leur est thermodynamiquement favorable. Peu de brins d’ADN matrice peuvent s’hybrider (se lier) avec leur brin complémentaire, ce qui empêcherait la fixation des amorces, car ces dernières sont bien plus courtes et en concentration bien plus importante. Expérimentalement, il est constaté que la PCR fonctionne même avec une phase d’hybridation avec une température supérieure de quelques degrés au Tm théorique des amorces, probablement parce qu’elles interagissent déjà avec les polymérases, qui stabiliseraient leur hybridation à l’ADN matrice.
Caractéristiques des amorces
- les séquences nucléotidiques des amorces doivent être spécifiques des séquences complémentaires de l'ADN simple-brin auxquelles elles vont s'apparier. La complémentarité parfaite n'étant par ailleurs pas obligatoire. De plus la spécificité de la séquence est importante dans la mesure où celle-ci ne doit pas pouvoir s'apparier à une autre séquence de l'ADN que l'on souhaite répliquer ;
- les séquences des amorces doivent être choisies de sorte à minimiser les possibilités d'appariement entre elles. De même chaque amorce est choisie pour ne pas pouvoir former une structure secondaire ;
- le procédé même de la PCR reposant, entre autres, sur des équilibres thermodynamiques, les amorces doivent avoir des températures de fusion le plus proche possible, autrement dit le rapport entre les bases AT et GC des deux amorces ne doit pas être trop différent.
Phase d’élongation (3 sur le schéma)
Cette étape (généralement 4 à 120 secondes à 72 °C) permet aux polymérases de synthétiser le brin complémentaire de leur ADN matrice à une température qui leur est optimale. Ce brin est fabriqué à partir des dNTPs libres présents dans le milieu réactionnel. La durée de cette étape dépend normalement de la longueur de l’amplicon.
Évolution de l'ADN au cours des quatre premiers cycles
Cycle no 1
- Lors de la phase 1, nous constatons que l’ADN initial a adopté une conformation « linéaire » (sans structure secondaire) et simple brin. Les amorces, les dNTPs et les polymérases sont en large excès et répartis de façon homogène dans la solution.
- Lors de la phase 2, une des amorces sens s’hybride avec sa séquence complémentaire sur le brin anti-sens (en magenta), une des amorces anti-sens se liant elle au brin sens (en orange). Deux polymérases peuvent alors interagir avec les deux complexes amorces/ADN matrice.
- Lors de la phase 3, les polymérases parcourent leur brin matrice de son extrémité 3’ vers son extrémité 5’ tout en synthétisant le brin complémentaire. Elles s’arrêteront à la fin du cycle, décrochées par la phase de dénaturation du cycle suivant. Les ADN néo-synthétisés sont donc précisément définis à leur extrémité 5’ mais pas à leur extrémités 3’ (parties noires). Les ADN sont alors bicaténaires sur une longueur plus ou moins importante.
À la fin de l’étape 3, nous avons alors deux brins d’ADN matrice et deux brins (un sens et un anti-sens) d’ADN précisément définis à leur extrémité 5’ uniquement.
Cycle no 2
Les trois phases se déroulent de la même manière qu’au cycle no 1, sauf que deux polymérases arrivées au bout de leur ADN matrice se décrochent spontanément. À la fin de la phase 3, nous obtenons tous les types d’ADN qui existeront lors de la PCR, soit :
- un brin d’ADN natif sens (A) ;
- deux brins d’ADN anti-sens précisément définis à leur extrémité 5’ uniquement (B) ;
- un brin d’ADN sens correspondant à l’amplicon, c'est-à-dire précisément défini à ses deux extrémités (C) ;
- deux brins d’ADN sens précisément définis à leur extrémité 5’ uniquement (D) ;
- un brin d’ADN anti-sens correspondant à l’amplicon, c'est-à-dire précisément défini à ses deux extrémités (E) ;
- un brin d’ADN natif anti-sens (F).
Cycle no 3
Idem au cycle 2. À la fin de la phase 3, nous observons 1 brin de type A et F, 3 de B et D, et 4 de C et E. Nous observons l’apparition de deux molécules d’ADN double brins C-E qui correspond à notre amplicon.
Cycle no 4
Idem au cycle 3. À la fin de la phase 4, nous observons 1 brin de type A et F, 4 de B et D, et 11 de C et E. Nous observons que l’amplicon devient la combinaison majoritaire.
Cycles no 5 et au-delà
Si nous avions augmenté le nombre de cycles, nous aurions obtenu le tableau suivant :
| cycles | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | n | ||
| type de molécules | A | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | |
| B | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | ||
| C | 0 | 0 | 1 | 4 | 11 | 26 | 57 | 120 | 247 | 502 | 1013 | 2036 | 4083 | 8178 | 16369 | 32752 | ||
| D | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | ||
| E | 0 | 0 | 1 | 4 | 11 | 26 | 57 | 120 | 247 | 502 | 1013 | 2036 | 4083 | 8178 | 16369 | 32752 | ||
| F | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||
| simple brin | nombre | 2 | 4 | 8 | 16 | 32 | 64 | 128 | 256 | 512 | 1024 | 2048 | 4096 | 8192 | 16384 | 32768 | 65536 | An à Fn |
| % amplicon | 0,00 | 0,00 | 25,00 | 50,00 | 68,75 | 81,25 | 89,06 | 93,75 | 96,48 | 98,05 | 98,93 | 99,41 | 99,68 | 99,83 | 99,91 | 99,95 | ||
| double brins | nombre | 1 | 2 | 4 | 8 | 16 | 32 | 64 | 128 | 256 | 512 | 1024 | 2048 | 4096 | 8192 | 16384 | 32768 | |
| % amplicon | 0,00 | 0,00 | 0,00 | 12,50 | 43,75 | 65,63 | 79,69 | 88,28 | 93,36 | 96,29 | 97,95 | 98,88 | 99,39 | 99,67 | 99,82 | 99,91 | ||










où An (Bn, etc.) est le nombre de molécules de type A (B, etc.) présentes après le nème cycle.
En analysant ce tableau, nous constatons que :
- les molécules d’ADN natif (A et F) ne sont pas dupliquées ;
- les molécules précisément définies à leur extrémité 5’ uniquement (B et D) augmentent de manière linéaire (de 1 par cycle sauf si l’ADN natif se dégrade) ;
- le nombre de molécules simple ou double brin augmente selon une exponentielle d’ordre 2 ;
- les molécules contenant la séquence exacte à amplifier (C et E) apparaissent dès le deuxième cycle et augmentent selon une suite arithmétique qui tend vers une exponentielle d’ordre 2 lorsque le nombre de cycles augmente. Notez que dès le dixième cycle, elles représentent près de 99 % de l’ensemble ;
- l'amplicon (les couples C-E) apparaît dès le troisième cycle et augmente selon une loi de même type que ses composants. Il représente près de 98 % des molécules au dixième cycle, 99,91 % au quinzième (zone où les ADNc issus d’ARNm fortement exprimés commencent généralement à devenir détectables).
Ces valeurs ont été obtenues en partant d’une seule molécule initiale d’ADN double brins, mais autrement, chaque matrice aurait subi le même processus. À un cycle donné, la quantité d’ADN dépend donc du nombre initial de matrices. En revanche, quelle que soit sa concentration initiale, il est théoriquement possible d’obtenir n’importe quelle quantité en ajustant le nombre de cycles. La PCR est donc régie théoriquement par la loi :
![[ADN]_{{cyclen}}=[ADN]_{{initiale}}xE^{n}](https://img.franco.wiki/i/bd2ed833a7c73b221b2f5a21acf8de15124b7b67.svg)
Mais la PCR est une réaction enzymatique complexe. Le produit est identique au substrat et peut venir inhiber l’enzyme, mais surtout, les réactifs secondaires (amorces, dNTP) peuvent commencer à manquer. La PCR ne peut donc avoir une loi d’amplification exponentielle que tant que l’ADN matrice est le seul facteur limitant. La réaction devient ensuite imprédictible et il est alors impossible de pouvoir comparer plusieurs échantillons entre eux sans un biais quantitatif plus ou moins significatif (voir l’article sur la PCR quantitative). Il est donc important de bien comprendre les différentes phases d’une cinétique de PCR.
Cinétique mesurable d'une PCR
Ce chapitre traite des cinétiques mesurables d'une PCR, car des limitations technologiques rendent inaccessibles les cycles précoces, et généralement une large portion de la partie exponentielle. Son profil apparent (mesurable) adopte plusieurs phases distinctes, plus ou moins développées en fonction de choix méthodologiques.
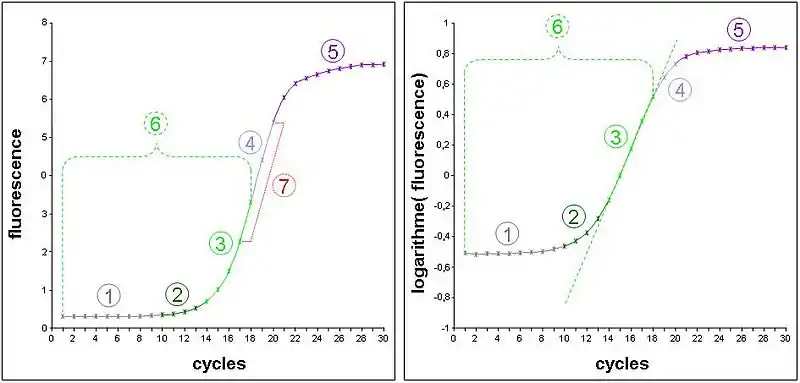
- Schéma de gauche : données expérimentales d’une cinétique de PCR mesurée sur un thermocycleur en temps réel. La fluorescence émise par un intercalant de l’ADN est proportionnelle à la quantité d’ADN présent (c'est-à-dire majoritairement l’amplicon). La courbe représente donc la cinétique d’amplification de l’ADN cycle par cycle.
- Schéma de droite : les valeurs de fluorescence précédentes ont été converties en logarithme décimal. Une cinétique de type exponentiel devrait donc être transformée en un segment de droite (segment quantifiable).
- 1) Bruit de fond de la fluorescence aspécifique du marqueur. Son niveau est très dépendant de la nature du marqueur et du soluté de l’échantillon.
- 2) Zone où la mesure de fluorescence est biaisée par le bruit de fond. Elle se distingue aisément sur le schéma de droite car elle adopte un profil d’exponentielle, alors que les valeurs sont déjà dans un repère semi-logarithmique.
- 3) Zone de la cinétique en phase exponentielle et mesurable sans biais. Elle adopte dans le schéma de gauche l’apparence d’un segment de droite nommé segment quantifiable. Remarquez que dans cet exemple expérimental, il ne fait que 5 cycles.
- 4) Zone de la cinétique où la polymérase devient un facteur limitant. L’accumulation d’amplicon se fait alors de manière constante et devient linéaire sur le schéma de gauche.
- 5) Phases d’amortissement puis de plateau de la cinétique dues à l’apparition de facteurs limitants non constants (dNTP, fluorophore, etc.), de dégradation de l’activité enzymatique, de la qualité de fluorescence…
- 6) Phase de la cinétique qui répondait réellement à une loi exponentielle, mais seule la zone 3 est réellement exploitable (sauf pour le maxima de dérivé seconde en PCR en temps réel).
- 7) Zone linéaire sur le schéma de gauche. Une erreur classique en PCR en point final était de déterminer cette zone grâce à une gamme de cycles pour tenter de faire une mesure quantitative de l’ADNc initial à ce niveau.
Efficacité de la PCR
NB : Le terme efficacité peut avoir deux significations selon les auteurs :
- les premiers désignent l’ordre de l’exponentielle. L’équation de la cinétique s’écrit donc : ;
- les seconds désignent la fraction de molécule d’ADN servant effectivement de matrice. L’équation devient alors : Cette deuxième formule facilite l’expression en pourcentage (en multipliant E par 100).
![[ADN]_{n}=[ADN]_{{initiale}}xE^{n}](https://img.franco.wiki/i/02a88bce8b96dda6284e28a90166c98f139e5996.svg)
![[ADN]_{n}=[ADN]_{{initiale}}x(1+E)^{n}](https://img.franco.wiki/i/ac1f273bfbfb381472a832cc7768c8d5ee02e69b.svg)
Les conclusions tirées de ces deux concepts (qui se déduisent directement l'un de l'autre) sont absolument identiques ; pour la discussion qui suit, on retiendra la première définition, majoritaire. Cette efficacité de PCR est un élément fondamental à prendre en compte pour obtenir une mesure quantitative ou établir un protocole de PCR multiplexe, mais elle est généralement négligeable pour un résultat qualitatif ou en PCR en point final.
Dans le cas d'un cycle théorique parfait, chaque brin matrice donne une et une seule copie complète de l’amplicon. Le nombre total de brins double donc à chaque étape, et l’efficacité théorique d’une réaction de PCR (notée E) est, dans ce cas, exactement égal à deux. Dans le graphique donnant le résultat dans un repère semi-logarithmique, cela se traduit par le fait que la pente de la partie rectiligne correspond à une progression idéale de log(2) par cycle (soit sensiblement 3 décibel par cycle).
L’avènement de la PCR en temps réel a mis en exergue que l’efficacité réelle de la réaction n’était pas toujours égale à deux. Cela signifie que sur l’ensemble des cycles considérés comme quantitatif (zone no 6 de la cinétique), dans les conditions réelles de la réplication, un brin peut ne pas donner une copie et une seule. Dans le graphique en repère semi-logarithmique, cela se traduit par une pente de la partie droite différente de la pente attendue, et généralement moins accentuée. La pente expérimentale est mesurable, et donne la valeur expérimentale de E. Le fait que cette partie est droite signifie que dans cet intervalle de concentration, le nombre moyen de copies réalisé par un brin est constant d'un cycle à l'autre (et en particulier, ne dépend pas des concentrations). Mais il n'y a pas, dans ce cas, « une copie et une seule par brin », mais un nombre différent.
La majorité des protocoles expérimentaux donne une efficacité de PCR entre 1,75 et 2. Deux phénomènes en sont les principales causes, et conduisent à une efficacité expérimentale inférieure ou égale à deux, parce que les réactions biochimiques n'étant pas toujours totales, certaines réplications échouent dans les conditions expérimentales réelles :
- tous les brins matrices ne sont pas forcément liés par un complexe amorce/polymérase lors de la phase d’hybridation (2), et cette liaison peut être réversible. La probabilité d’amorçage (ou de décrochage) peut être influencée par la température, la longueur et la séquence de l’amorce, sa composition en nucléotides naturels ou modifiés, sa concentration dans la solution, la composition ionique de cette dernière, l’auto-hybridation entre les amorces ou la compétition possible d’ADN non cible (par exemple un pseudo-gène) ;
- toutes les synthèses (3) ne sont pas forcément complètes, notamment si la phase d’élongation est trop courte. Un brin incomplet à son extrémité 3’ ne peut pas servir de matrice, car l’amorce complémentaire ne peut pas se lier à lui.
Inversement, certaines sources considèrent peut également être supérieure à deux (ce qui suppose qu'un brin a une probabilité significative de réaliser deux ou plusieurs synthèses pendant un cycle), et serait acceptable jusqu’à 2,3. Elle peut s'expliquer de deux manières :
- la température et la durée optimale de la phase d'élongation (3) peut conserver une cinétique non nulle à la fois pour la réaction de dénaturation (1) et celle d'amorçage (2). Donc pour chacune de ces matrices, après qu'un complexe amorce/polymérase s’est fixé une première fois, une synthèse complète a été effectuée, l’ADN double brin peut se déshybridé (même à une température défavorable), et un second complexe amorce/polymérase s’est fixé à une température probablement défavorable (étape d’élongation) et qu’une seconde synthèse complète peut recommencer (voire une troisième). Dans ce cas, une part significative (jusqu’à 30 %) des brins d’ADN servent plusieurs fois de matrice pendant un cycle, et cette proportion (qui ne dépend que des cinétiques des réactions) est conservée pendant toute la phase exponentielle ;
- un biais expérimental (dans un facteur de dilution par exemple) ou d’analyse mathématique (généralement commis par l’utilisation inappropriée d’outil d’optimisation présent dans le logiciel du thermocycleur) a été commis. Ces deux types de biais ont été plusieurs fois constatés.
Deux écoles s’affrontent alors, avec des arguments expérimentaux à l’appui. L’une considère que cette efficacité est une constante pour chaque amplicon dans un protocole expérimental donné. L’autre estime qu’elle varie toujours significativement et qu’elle nécessite d’être constamment remesurée. Il convient de noter qu’il est très difficile de savoir si une variation d’une efficacité de PCR observée vient de la nature même de cette méthode, d’une variation dans le protocole expérimental (manque de reproductibilité des réactifs ou de la manipulation) ou d’une variation dans l’acquisition des données (variations de fluorescence, canaux de lecture différents, biais dans l’analyse mathématique). Il est également très ardu d’être certain que l’efficacité utilisée (mesurée à chaque expérience ou non) est bien celle qui a eu cours dans l’échantillon à calibrer (voir la PCR quantitative).
Techniques associées à la PCR
Les sigles et les noms en anglais sont donnés entre parenthèses.
PCR multiplexe
La PCR multiplexe (multiplex PCR) est un protocole destiné à amplifier plus d’un amplicon à la fois, par l'utilisation d'au moins trois amorces par réaction de PCR. Les produits de PCR ne seront alors compétitifs que pour la polymérase, les dNTP et, éventuellement, le marqueur d’ADN. Il est également possible d’amplifier différents types d’ADN reconnus par un même couple d’amorces, tels les mimics. La PCR multiplexe peut se faire en point final (les produits de PCR étant usuellement différenciés par leur taille ou la présence d’un site de restriction) ou en temps réel (chaque produit étant mesuré par une sonde spécifique couplée à un fluorophore dont le spectre d’émission est différent des autres). Ses applications qualitatives sont nombreuses (détection de souche virale, de mutations…) mais son aspect quantitatif ne fait pas l’unanimité, malgré de fortes pressions industrielles pour ce marché très lucratif.
Méta-PCR
Meta-PCR est une méthode qui permet de donner une molécule d'ADN synthétique comprenant n'importe quelle combinaison d'ADN amplifiable par PCR dans n'importe quel ordre. Elle est effectuée dans un seul tube et se compose de deux différentes étapes de la PCR séparées par un cycle de dégel pour éliminer l'activité résiduelle de la polymérase. La Meta-PCR peut être utilisée pour accroître le débit des méthodes de numérisation et balayage des mutations.
PCR emboîtée ou Nested PCR
La PCR emboîtée, PCR gigogne ou PCR nichée (Nested PCR) est une PCR en deux étapes successives, avec deux couples d’amorce différents, le second liant des séquences situées à l’intérieur du premier amplicon. Cette technique était initialement utilisée pour réduire le risque de contamination (le produit final devait pouvoir interagir avec deux couples d’amorces, donc deux niveaux de spécificité). Elle est maintenant très utilisée par les virologues travaillant sur les virus à ARN qui peuvent avoir une haute mutabilité. Le premier couple d’amorces est conçu pour pouvoir accrocher les quelques parties stables du génome viral, le deuxième pour identifier le sous-type. Elle permet aussi une meilleure sensibilité du résultat.
PCR asymétrique
C'est l'amplification par PCR en présence d'une faible quantité d'une des amorces. Elle permet le séquençage direct des fragments amplifiés. Pendant les 20 à 25 premiers cycles, l'ADN double brin est généré, jusqu'à épuisement de l'amorce limitante et de l'ADN simple brin est produit pendant les 5 à 10 derniers cycles. Les rapports d'amorces utilisés sont de 50 pmole/1-5 pmoles/100 µL (1-3 pmoles simple brin après 30 cycles).
PCR à asymétrique thermique entrelacée
La PCR à asymétrique thermique entrelacée (TAIL-PCR ou Thermal asymmetric interlaced PCR) est un protocole complexe alliant les principes de la PCR emboîtée, de la PCR asymétrique par le Tm des amorces, la succession de plusieurs types de cycle favorisant l’hybridation de telle ou telle amorce, et des amorces dégénérées. Le but est d’obtenir un amplicon final spécifique d’une séquence d’ADN dont seule une extrémité est connue généralement dans l’optique de séquencer la partie inconnue. Cette méthode est très utilisée pour la marche sur chromosome ou la caractérisation des séquences variables des immunoglobulines.
PCR en gradient de température
Il s’agit d’une technique aidant à la mise au point d’un nouveau protocole de PCR. Elle nécessite des thermocycleurs capables d’assurer des températures différentes, pour une même étape, aux différents échantillons. Elle est surtout utilisée pour optimiser l’étape d’hybridation, notamment en PCR multiplexe.
PCR quantitative
PCR semi-quantitative
La PCR semi-quantitative est basée sur l'interruption de la PCR en plusieurs cycles qui correspondent à la phase stationnaire (la quantité d'ADN augmente très doucement car il y a peu d'ADN matrice), la phase exponentielle (croissance rapide) et au plateau (diminution de la quantité de réactifs). Pour un échantillon, il est possible d'estimer la quantité initiale d'ADN si l'on dispose d'un échantillon où la quantité d'ADN est connue (étalon). Leur amplification par PCR, l'arrêt de la réaction entre le 20e et le 30e cycle PCR et la comparaison entre les intensités lumineuses des produits PCR marqués au BET permettent d'estimer la quantité initiale d'ADN. Pour deux échantillons, il est possible de comparer leur quantité d'ADN initiale en arrêtant la PCR avant le plateau de la PCR (<30 cycles).
PCR en temps réel ou PCR quantitative
La PCR en temps réel (Real-time PCR) est une révolution dans l’utilisation de la PCR, cette technique consiste à mesurer la quantité d’ADN polymérisé à chaque cycle (temps réel) grâce à un marqueur fluorescent. Elle permet par son principe de faire des mesures quantitatives (expliquant l'appellation PCR quantitative, qPCR) mais elle nécessite des thermocycleurs particuliers. Il ne faut surtout pas la confondre avec la RT-PCR (Reverse Transcription PCR), on préférera donc les appellations PCR quantitative ou qPCR. Certaines expériences en PCR compétitive ou PCR radioactive permettent l'obtention de mesure quantitative exploitable.
PCR en point final
La PCR en point final (end point PCR) est un terme qui est apparu en opposition à la PCR en temps réel. Il désigne toutes les tentatives de quantification à partir du produit final d’une réaction de PCR.
PCR par essais
La PCR par essais (Touchdown PCR) est un protocole utilisé pour amplifier de l'ADN faiblement représenté et/ou subissant une compétition sur leurs amorces par des produits de pseudo-gène. Il consiste à avoir une température d’hybridation très haute lors des premiers cycles afin d’assurer une forte stringence et donc une amplification spécifique. Une fois que la séquence d'intérêt devient majoritaire vis-à-vis de ses compétiteurs, la température d’hybridation est progressivement abaissée afin d’assurer une meilleure efficacité de PCR. L’efficacité n’étant pas constante tout au long de la réaction, il n'est pas possible, à ce jour, de pouvoir obtenir un résultat quantitatif sans biais.
PCR sur colonie
La PCR sur colonie (Colony PCR) est un protocole qui permet d'amplifier de façon simple de l'ADN de micro-organisme (bactéries, archées ou levures) en inoculant directement la colonie dans le milieu réactionnel de la PCR. Durant les premières étapes de dénaturation, les cellules sont lysées et leur ADN est libéré dans le milieu réactionnel. L'ADN ainsi libéré peut alors servir de matrice.
La PCR sur colonie est utilisée notamment pour vérifier l'efficacité d'une transgénèse. Elle fut très utilisée lors de la construction des BAC dans les grands programmes de séquençage.
La PCR sur colonie est largement utilisée en recherche microbiologique afin de caractériser sur le plan phylogénétique les souches étudiées.
ep-PCR (error prone PCR)
L'ep-PCR permet d'introduire des erreurs dans la séquence copiée par la polymérase. Cette technique peut être utilisée par exemple pour créer un grand nombre de mutants qui seront ensuite sélectionnés (évolution dirigée). La polymérase utilisée (la Taq polymerase est naturellement moins fidèle que les polymérases classiques en PCR) et des conditions particulières (fortes concentrations en MgCl2 ou ajout de MnCl2) permettent d'augmenter le taux d'erreurs[6].
Après transcription inverse
RT-PCR
La RT-PCR (reverse transcription‐polymerase chain reaction) est une technique qui associe une transcription inverse (RT) suivie d’une PCR. Elle synthétise le brin complémentaire d’un ARN avec des désoxyribonucléotides en utilisant une ADN polymérase ARN dépendante (transcriptase inverse). Cet ADNc est généralement destiné à être amplifié par PCR (l'ADNc étant plus stable, il permet plus de liberté que les ARN pour les analyses suivantes).
Utilité : elle sert au diagnostic clinique, via des mesures quantitatives par PCR en temps réel (étude des charges virales en virus à ARN) et via des mesures qualitatives (analyse du produit de l‘expression des gènes)[7]. Elle peut aussi faciliter le séquençage de grands gènes[7].
La transcription inverse reste délicate (faible reproductibilité ; fragilité de l‘ARN, fréquentes contamination par de l‘ADN. Une amplification spécifique est parfois nécessaire (si le milieu contient des séquences homologues en quantités très supérieures à celles de la séquence d‘intérêt). Le choix des amorces de PCR et le respect de standards internes ou externes ont souvent une grande importance[7].
RT-PCR en une étape
La RT-PCR en une étape (single step RT-PCR) est un protocole mélangeant les réactifs de RT et de PCR afin que les deux étapes puissent se faire sans avoir à ouvrir le tube. Cela permet de réduire le risque de contamination ou d’inversion d’échantillon mais il est plus difficile d’optimiser le milieu réactionnel pour chaque étape. Cela induit en outre un risque de biais pour normaliser l’étape de RT car cela implique d'utiliser la PCR multiplexe.
RT-PCR in situ
La RT-PCR in situ est une méthodologie qui consiste à réaliser la RT-PCR non pas sur des molécules en solution mais sur des coupes histologiques. Bien que les résultats ne soient que semi-quantitatifs, ils apportent en outre une information sur la localisation des transcrits dans le tissu.
RT-PCR quantitative
La RT-PCR quantitative (qRT-PCR) est une technique destinée à pouvoir quantifier un type d’ARN initialement présent dans un échantillon. Ce terme désigne l'utilisation de deux techniques successivement, une Transcription inverse suivie d’une PCR en temps réel. Objectif principal de la plupart des biologistes, elle soulève de vives polémiques quant à l’utilisation de calibrateur externe ou interne (gène de ménage). À l’exception de complexes protocoles utilisant des calibrateurs externes homologues compétitifs, elle ne permet qu’une quantification relative.
RT-PCR sur une cellule
La RT-PCR sur une cellule (single-cell RT-PCR) est une méthodologie qui permet d’étudier les transcrits d’une cellule unique, obtenue par patch-clamp, microdissection laser ou triage de cellules par activité de fluorescence (FACS en anglais pour Fluorescence activated cell sorting). Cette précision peut être nécessaire lorsque le tissu n’est pas homogène (cellules tumorales, feuillets cellulaires bien différenciés, etc.) mais la quantification va devenir moins précise à cause d’une amplification des effets stochastiques et nécessite donc une multiplication des mesures.
PAN-AC
Une autre technique d'amplification isotherme : « polymérisation des acides nucléiques pour apoptose contrôlée ». Les auteurs prétendent que cette technique pourrait être mise en œuvre dans des cellules vivantes, et ainsi soigner diverses pathologies (HIV, malaria, cancers, etc.)[8] mais ces affirmations n'ont été corroborées par aucune autre équipe pour le moment.
TP-PCR
La TP-PCR (TP pour Triplet Repeat primed), une variante complexe de la PCR, a été mise au point par Jon Warner en 1996 et est utilisée dans les amplifications des gènes comportant des triplets répétés, comme dans le cas de la dystrophie myotonique de Steinert. La combinaison PCR-séquençage ne peut pas être utilisée dans ces cas car la Taq polymérase fait des erreurs de réplication.
Amplification hélicase-dépendante
L'amplification hélicase-dépendante (helicase-dependent amplification, HDA) est une technique récente proche de la PCR, où la déshybridation induite par la température est assurée par une hélicase.
Applications
Pour les maladies infectieuses
La PCR permet un diagnostic rapide et hautement spécifique des maladies infectieuses causées par des bactéries ou des virus[9]. La PCR permet également l'identification de micro-organismes non cultivables ou à croissance lente tels que les mycobactéries, les bactéries anaérobies ou les virus à partir d'essais de culture tissulaire et de modèles animaux. La base des applications de diagnostic PCR en microbiologie est la détection d'agents infectieux et la discrimination des souches non pathogènes des souches pathogènes en raison de gènes spécifiques [9],[10].
La caractérisation et la détection des organismes infectieux ont été révolutionnées par la PCR de la manière suivante :
Le virus de l'immunodéficience humaine (ou VIH) est une cible difficile à trouver et à éradiquer. Les premiers tests d'infection reposaient sur la présence d'anticorps dirigés contre le virus se trouvant dans la circulation sanguine. Cependant, les anticorps n'apparaissent que plusieurs semaines après l'infection, les anticorps maternels masquent l'infection d'un nouveau-né et les agents thérapeutiques pour lutter contre l'infection n'affectent pas les anticorps. Des tests de PCR ont été développés pour pouvoir détecter des charges virales faibles de l'ordre d'un génome viral parmi l'ADN de plus de 50 000 cellules hôtes[11]. Les infections peuvent être détectées plus tôt, le sang donné peut être dépisté directement pour le virus, les nouveau-nés peuvent être immédiatement testés pour l'infection et les effets des traitements antiviraux peuvent être quantifiés.
Certains organismes pathogènes, comme celui de la tuberculose, sont difficiles à prélever sur les patients et lents à se développer en laboratoire. Des tests basés sur la PCR permettent de détecter un petit nombre d'organismes pathogènes (vivants ou morts) dans des échantillons. Une analyse génétique détaillée peut également être utilisée pour détecter la résistance aux antibiotiques, permettant un traitement immédiat et efficace. Les effets d'une thérapie antibiotique peuvent également être immédiatement évalués.
La propagation d'un organisme pathogène à travers des populations d'animaux domestiques ou sauvages peut être surveillée par des tests PCR. Dans de nombreux cas, l'apparition de nouveaux sous-types virulents peut être détectée et surveillée. Les sous-types d'un organisme qui étaient responsables d'épidémies antérieures peuvent également être déterminés par PCR.
L'ADN viral peut être détecté par PCR. Les amorces utilisées doivent être spécifiques aux séquences ciblées dans l'ADN d'un virus, et la PCR peut être utilisée pour des analyses diagnostiques ou le séquençage d'ADN du génome viral. La sensibilité élevée de la PCR permet la détection du virus peu de temps après l'infection et avant même le début des symptômes de la maladie[9]. Une détection aussi précoce peut donner aux médecins un choix de traitement plus important. La quantité de virus (charge virale) chez un patient peut également être quantifiée par des techniques de quantification d'ADN basées sur la PCR. Par exemple, une variante de la PCR (RT-PCR) est utilisée pour détecter le génome viral de la Maladie à coronavirus 2019[12].
Des maladies telles que la coqueluche sont causées par la bactérie Bordetella pertussis. Cette bactérie provoque une grave infection respiratoire aiguë qui affecte divers animaux et humains et entraîne la mort de nombreux jeunes enfants. La toxine coquelucheuse est une exotoxine protéique qui se lie aux récepteurs cellulaires par deux dimères et réagit avec différents types cellulaires tels que les lymphocytes T qui jouent un rôle dans l'immunité cellulaire[13]. La PCR est un outil de test important qui peut détecter des séquences dans le gène de la toxine coquelucheuse. Parce que la PCR a une sensibilité élevée à la toxine et un délai d'exécution rapide, elle est très efficace pour diagnostiquer la coqueluche par rapport à la culture[14].
Autres techniques
- ARDRA
- Hybridation in situ
- LAMP
- Microarray
- NASBA
- Northern blot
- RACE-PCR
- RNAse Protection Assay (RPA)
- Southern blot
- TMD
- ADN branché
- LCR
- PAN-AC (voir : http://www.lab-rech-associatives.com/pages/agi.php )
- MALBAC
Notes et références
- ↑ Commission d’enrichissement de la langue française, « amplification en chaîne par polymérase », sur FranceTerme, ministère de la Culture.
- ↑ « amplification en chaîne par polymérase », Grand Dictionnaire terminologique, Office québécois de la langue française (consulté le ).
- ↑ [vidéo] Principe de la PCR (qualité et marche en avant) sur YouTube, (consulté le ).
- ↑ « Comment fonctionnent les tests de dépistage du Covid-19 ? », Ens-paris-Saclay, (lire en ligne).
- 1 2 Initiation à la biologie moléculaire ; Module : Amplification de gène, Fondation Mérieux
- ↑ Cirino et al., « Generating Mutant Libraries Using Error-Prone PCR »], Methods in Molecular Biology, volume 231, 2003.
- 1 2 3 Cavé, H., Acquaviva, C., Bièche, I., Brault, D., Fina, F. D. F. F., Loric, S., ... & Tuffery, S. (2003). La RT‐PCR en diagnostic clinique. In Annales de biologie clinique (Vol. 61, No. 6, pp. 635-644).
- ↑ (en) F. David et E. Turlotte, « An Isothermal Amplification Method », Comptes-Rendus de l'Académie des Sciences, vol. 321, , p. 909-914
- 1 2 3 H Cai, Caswell JL et Prescott JF, « Nonculture Molecular Techniques for Diagnosis of Bacterial Disease in Animals: A Diagnostic Laboratory Perspective », Veterinary Pathology, vol. 51, no 2, , p. 341–350 (PMID 24569613, DOI 10.1177/0300985813511132)
- ↑ (en) Salis AD, Real-Time PCR : Current Technology and Applications, Norfolk, Caister Academic Press, , 284 p. (ISBN 978-1-904455-39-4), « Applications in Clinical Microbiology »
- ↑ S. Kwok, D. H. Mack, K. B. Mullis, B. Poiesz, G. Ehrlich, D. Blair, A. Friedman-Kien et J. J. Sninsky, « Identification of human immunodeficiency virus sequences by using in vitro enzymatic amplification and oligomer cleavage detection », Journal of Virology, vol. 61, no 5, , p. 1690–4 (PMID 2437321, PMCID 254157, DOI 10.1128/jvi.61.5.1690-1694.1987)
- ↑ « Coronavirus: il viaggio dei test », sur Istituto Superiore di Sanità
- ↑ Horst Finger et Carl Heinz Wirsing von Koenig, Medical Microbiology, Galveston (TX), University of Texas Medical Branch at Galveston, , 4e éd. (ISBN 978-0-9631172-1-2, PMID 21413270, lire en ligne)
- ↑ Sylvia H. Yeh et ChrisAnna M. Mink, Netter's Infectious Diseases, , 11–14 p. (ISBN 978-1-4377-0126-5, DOI 10.1016/B978-1-4377-0126-5.00003-3), « Bordetella pertussis and Pertussis (Whooping Cough) »
Voir aussi
Bibliographie
- Élyse Poitras et Alain Houde, « La PCR en temps réel: principes et applications », Reviews in Biology and Biotechnology (Canada), vol. 2, no 2, . p. 2-11
- Initiation à la biologie moléculaire ; Module : Amplification de gène, Fondation Mérieux
Articles en anglais
- (en) Baltimore D. RNA-dependent DNA polymerase in virions of RNA tumor viruses. 1970. Nature; 226, 1209–1211
- (en) Birch DE. Simplified hot start PCR. 1996. Nature; 381:445–6.
- (en) Friedberg EC. The eureka enzyme: the discovery of DNA polymerase. Nat Rev Mol Cell Biol. 2006 Feb;7(2):143-7.
- (en) Higuchi R, Dollinger G, Walsh PS, Griffith R. « Simultaneous amplification and detection of specific DNA sequences » Biotechnology. 1992;10:413–7.
- (en) Holland PM, Abramson RD, Watson R, Gelfand DH. « Detection of specific polymerase chain reaction product by utilizing the 5’–3’ exonuclease activity of Thermus aquaticus DNA polymerase » Proc Natl Acad Sci USA 1991;88:7276–80].
- (en) Kornberg, A., Lehman, I. R., Bessman, M. J & Simms, E. S. « Enzymatic synthesis of desoxyribonucleic acid ». 1956. Biochim. Biophys. Acta 21, 197–198
- (en) Lay MJ, Wittwer CT. « Real-time fluorescence genotyping of factor V Leiden during rapid-cycle PCR ». 1997. Clin Chem ;43:2262–7.
- (en) Liu YG, Whittier RF. « Thermal asymmetric interlaced PCR: automatable amplification and sequencing of insert end fragments from P1 and YAC clones for chromosome walking ». 1995. Genomics. Feb 10;25(3):674-81.
- (en) Mullis K, Faloona F, Scharf S, Saiki R, Horn G, Erlich H. Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harb Symp Quant Biol. 1986;51 Pt 1:263-73.
- (en) Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, Mullis KB, Erlich HA. Primer-directed enzymatic amplification of DNA with a thermostable DNA polymerase. Science. 1988 Jan 29;239(4839):487-91.
- (en) Stoflet ES, Koeberl DD, Sarkar G, Sommer SS. Genomic amplification with transcript sequencing. Science. 1988 Jan 29;239(4839):491-4.
- (en) Temin HM & Mizutani S. RNA-dependent DNA polymerase in virions of Rous sarcoma virus. 1970. Nature 226, 1211–1213 (1970).
- (en) Watson JD & Crick FC. « A structure for deoxyribose nucleic acid », Nature. 1953 171, 737–738
- (en) Warner JP et al. A general method for the detection of large CAG repeat expansions - Journal of Medical Genetics, December 1996, Vol. 33, No 12, p. 1022 – 1026
- (en) Wittwer CT, Herrmann MG, Moss AA, Rasmussen RP. Continuous fluorescence monitoring of rapid cycle DNA amplification. 1997. Biotechniques;22:130–1, 134–8.
Articles connexes
- Techniques de biologie moléculaire
- Liste d'abréviations de biologie cellulaire et moléculaire
- Téléportation de l'ADN
- PCR in silico
- PCR par essais
- Maladie à coronavirus 2019 alias Covid-19
Liens externes
- Animation PCR : principe de la PCR et de la PCR en temps réel et comparaison de ces techniques
- « Animation »(Archive.org • Wikiwix • Archive.is • Google • Que faire ?)
- « Animation PCR : principe de la PCR et de la PCR en temps réel et comparaison de ces techniques »(Archive.org • Wikiwix • Archive.is • Google • Que faire ?)
- « Animation (université de Lyon) »(Archive.org • Wikiwix • Archive.is • Google • Que faire ?)
- Animation PCR - (Flash)