ساركومة يوينغ أو ساركوما إيوينغ (Ewing's sarcoma) هو أحد الأورام الخبيثة ذات الخلايا المستديرة الزرقاء الصغيرة، وهو مرض نادر توجد فيه الخلايا السرطانية في العظام أو في الأنسجة الرخوة. مناطق الجسم الأكثر شيوعًا لحدوثه هي الحوض وعظم الفخذ والعضد والأضلاع والترقوة.
| ساركومة يوينغ | |
|---|---|
 | |
| معلومات عامة | |
| الاختصاص | علم الأورام |
| من أنواع | ساركومة، وسرطان العظام، وعائلة يوينغ للأورام، وساركوما العظم |
| الإدارة | |
| أدوية | |
وبما أن الموضع الجيني المسؤول عن نسبة كبيرة من ساركومة يوينغ والأورام العصبية الابتدائية مشترك، يتم تجميعها أحيانًا معًا في فئة تُعرف باسم عائلة يوينغ للأورام.[1]
يحدث ساركومة يوينغ في كثير من الأحيان في المراهقين والشباب، مع نسبة الذكور/الإناث 1.6:1.[2]
وعلى الرغم من تصنيفه عادةً على أنه ورم في العظام، يمكن أن يكون لساركومة يوينغ خصائص ذات أصل من الأديم المتوسط والأديم الظاهر، مما يجعل من الصعب تصنيفه.[3]
كان جيمس يوينغ (1866-1943) أول من وصف الورم، مؤكدًا أن المرض منفصل عن سرطان الغدد الليمفاوية وأنواع السرطان الأخرى المعروفة في ذلك الوقت.[4][5]
الأعراض
ساركومة يوينغ أكثر شيوعًا في الذكور (1.6 ذكور: 1 إناث) وعادةً ما يظهر في مرحلة الطفولة أو بداية مرحلة البلوغ، خاصةًً بين سن 10 و20 من العمر. يمكن أن يحدث في أي مكان في الجسم، ولكن الأكثر شيوعًا في الحوض والعظام، وخاصةً حول لوحات النمو. والمنطقة الأكثر شيوعًا هي جدل عظم الفخذ، تليها الساق والعظم العضدي. يكون ثلاثون في المئة منه منبثة عند الفحص، وعادةً ما يعاني المرضى من آلام شديدة في العظام.
تشمل العلامات والأعراض: الحمى المتقطعة وفقر الدم وزيادة عدد الكريات البيضاء وزيادة معدل الترسيب وأعراض أخرى من أمراض الجهاز التنفسي. أيضًا قد يحدث ألم شديد اعتمادًا على نوع وتطور وموقع الورم.
وفقًا لأمانة أبحاث سرطان العظام، الأعراض الأكثر شيوعًا هي: الألم الموضعي والتورم وآلام متفرقة في العظام بشدة متغيرة. ومن المرجح أن يكون التورم مرئيًا إذا كان الورم موجودًا على عظم قريب من سطح الجسم، ولكن عندما يحدث في أماكن أخرى أعمق في الجسم مثل الحوض، قد لا يكون مرئيًا.[6]
الأسباب
يمكن أن يُحوِّل التبادل الوراثي بين الكروموسومات الخلايا إلى سرطانية. معظم الحالات من ساركومة يوينغ (85٪) هي نتيجة للنقل بين الكروموسومات 11 و22، حث يندمج الجين EWS من الكروموسوم 22 إلى الجين FLI1 من الكروموسوم 11.[7] هناك انتقالات أخرى مثل (21,22)[8] و(7، 22).[9] وتكون خلايا ساركوما إوينغ إيجابية لCD99 وMIC2، وسلبية لCD45.[10]
التشخيص

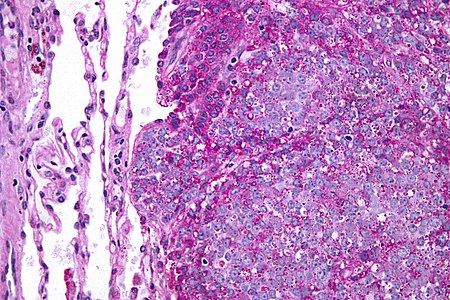
يعتمد التشخيص في الأساس على نتائج علم الأنسجة والكيمياء النسيجية المناعية وعلم الأمراض الجزيئي.
ساركومة يوينغ هو ورم من الخلايا الصغيرة الزرقاء المستديرة التي عادة ما تكون ذات سيتوبلازم نقي باستخدام صبغة الهيماتوكسيلين والإيوسين نتيجة وجود الجليكوجين. يمكن إيضاح وجود الجليكوجين بالنتيجة الإيجابية لصبغة PAS والنتيجة السلبية لصبغة دياستاز PAS. والملون المناعي المميز هو CD99، والذي يحدد غشاء الخلية بشكل كبير. يتم إثبات النتائج المورفولوجية والمناعية مع انتقال الكروموسومات الذي يحدث عادة. أكثر التكرارات شيوعًا التي تظهر في حوالي 90٪ من حالات ساركوما إوينغ هي الانتقال (11؛ 22) (q24؛ q12)،[11][12] الذي يولد عامل النسخ الشاذ من خلال دمج جين EWSR1 مع الجين FLI1.[13]
التشخيص التفريقي المَرَضي هو مجموعات الأورام ذات الخلايا الصغيرة الزرقاء المستديرة، والتي تشمل سرطان الغدد الليمفاوية والساركومة العضلية المخططة السنخية والأورام الصلدة (من النسيج الليفي) وغيرهم.
التشخيص التفريقي
تشمل الكيانات الأخرى ذات الأعراض المماثلة التهاب العظم والساركوما العظمية (وخاصة الساركومة العظمية متوسعة الشعيرات) والورم الحبيبي اليوزيني. الأورام اللينة الأنسجة مثل الساركومة غير المتمايزة متعددة الأشكال (ورم المنسجات الليفية الخبيث) -التي تفتت العظام المجاورة- قد يكون لها مظهر مماثل.
نتائج التصوير


على الأشعة التقليدية، العَرَض العظمي الأكثر شيوعًا هو الآفة نافذة التحلل مع تفاعل سمحاقي. والوصف الكلاسيكي للتفاعل السمحاقي مع هذه الآفة في شكل طبقات. تضيف الأفلام العادية معلومات قيمة في التقييم الأولي أو التحري. المنطقة الواسعة من الانتقال (على سبيل المثال: النفاذية) هي السمة الأكثر فائدة في الأشعة للتمييز بين الآفات الحميدة والخبيثة. ينبغي استخدام التصوير بالرنين المغناطيسي بشكل روتيني في فحص الأورام الخبيثة. فهي تُظهر كامل نطاق الأنسجة العظمية واللينة وتربط الورم إلى الهياكل التشريحية القريبة الأخرى (مثل الأوعية). تباين الغادولينيوم ليس ضروريًا لأنه لا يعطي معلومات إضافية عن الدراسات غير المتقابلة، على الرغم من جدال بعض الباحثين الحاليين بأن التصوير بالرنين المغناطيسي الديناميكي المعزز على النقيض من شأنه أن يساعد على تحديد مقدار النخر داخل الورم، وبالتالي يساعد في تحديد الاستجابة للعلاج قبل الجراحة.
يمكن أيضًا استخدام التصوير المقطعي المحوري لتحديد مدى خروج الورم عن العظام، وخاصة في الجمجمة والعمود الفقري والأضلاع والحوض. ويمكن استخدام كل من التصوير بالرنين المغناطيسي والتصوير المقطعي المحوري لمتابعة الاستجابة للإشعاع و أو العلاج الكيميائي. يمكن أيضًا استخدام تصوير العظام باستخدام الفطائر العظمية لمتابعة استجابة الورم للعلاج.
في مجموعة الأورام الخبيثة المستديرة الصغيرة والتي تشمل ساركومة يوينغ وسرطان الغدد الليمفاوية العظمية وسرطان العظام في الخلايا العظمية الصغيرة، قد تظهر القشرة طبيعية بالإشعاع بينما تحدث النفاذية عبر قنوات هافيرسيان. قد تكون هذه الأورام مصحوبة بكتلة كبيرة من الأنسجة الرخوة ولا يحدث تدمير للعظام. وغالبًا لا تُظهِر الصور الشعاعية أي علامات لتدمير القشرة.
ومن الناحية الإشعاعية، تتمثل ساركومة يوينغ كمناطق شفافة "متخربة بالعث" في النخاع وتآكل في القشرة.
العلاج
يحتاج جميع المرضى تقريبًا إلى العلاج الكيميائي متعدد الأدوية (متضمِّنًا إفوسفاميد وإيتوبوسيد)، وكذلك التحكم في المرض بالجراحة و/أو الإشعاع. وكون العلاج المكثف ضروريًا لأن غالبًا جميع المرضى الذين يظهر عليهم ورم في منطقة محددة في وقت التشخيص يكون لديهم في الواقع مرض منبث غير ظاهر.
غالبًا ما يتكون العلاج من العلاج الكيميائي المساعد، والذي قد يشمل فينكريستين ودوكسوروبيسين وسيكلوفوسفاميد مع (إفوسفاميد و إيتوبوسيد).[14] بعد حوالي ثلاثة أشهر من العلاج الكيميائي، يتم استئصال الورم المتبقي جراحيًا أو بالإشعاع أو كليهما،[15] وقد يشمل الاستئصال الجراحي بتر الأطراف. ويمكن إجراء الاستئصال الكامل في وقت الخزعة إذا تأكد أن الورم خبيث في وقت الفحص.
تختلف مدة العلاج اعتمادًا على موقع ومرحلة المرض عند التشخيص. قد يكون العلاج الكيميائي الكلي قصير مثل ستة علاجات في دورات لمدة 3 أسابيع، ولكن يخضع معظم المرضى للعلاج الكيميائي لمدة 6-12 شهرًا والعلاج الإشعاعي لمدة 5-8 أسابيع. ويُستخدم العلاج الإشعاعي للورم في موقع محدد. للورم خاصية فريدة من نوعها لكونه حساس للغاية للإشعاع، ولكن العيب الرئيسي هو أنه يتكرر بشكل كبير بعد بعض الوقت.
الحفاظ على الخصوبة
في النساء، قد يضر العلاج الكيميائي بالمبايض ويسبب العقم. للحفاظ على الحمل في المستقبل، يمكن للمرأة الحفاظ على البويضات أو الأنسجة المبيضية عن طريق حفظ البيض بالتبريد أو حفظ أنسجة المبيض بالتبريد قبل البدء في العلاج الكيميائي.[16] إلا أن هذا قد يُعيد السرطان عند إعادة اندماج الأنسجة المبيضية. إذا تم تنفيذه، يجب فحص الأنسجة المبيضية لتتبع الآثار الخبيثة على كل من المستويات المرضية والجزيئية قبل تطعيم الأنسجة المحفوطة تبريديًا.
تقدُّم المرض
محاولات التدريج لتمييز المرضى ذوي الأورام المحددة من أولئك الذين يعانون من ورم منبث.[17] والأكثر شيوعا يحدث الانبثاث في الصدر والعظام و/أو نخاع العظام. وتشمل المواقع الأقل شيوعًا الجهاز العصبي المركزي والغدد الليمفاوية.
معدل البقاء على قيد الحياة لمدة خمس سنوات في حالة الورم المحدد هو 70٪ إلى 80٪ عند استخدام العلاج الكيميائي.[18] قبل استخدام العلاج الكيميائي متعدد الأدوية، كان البقاء على قيد الحياة لمدى الطويل أقل من 10٪. وقد أدى تطوير العلاج متعدد التخصصات مع العلاج الكيميائي والإشعاع والجراحة إلى زيادة المعدلات الحالية للبقاء على قيد الحياة على المدى الطويل في معظم المراكز السريرية إلى أكثر من 50٪. وتشير بعض المصادر إلى أنها 25-30٪.[19]
أظهرت البحوث بأثر رجعي في المرضى بقيادة إدريس بناني بيتي (جمعية علم وراثة السرطان) أن اثنين من مستقبلات كيموكين CXCR4 وCXCR7، يمكن أن تستخدم عوامل التكهن الجزيئي. المرضى الذين لديهم مستويات منخفضة من مستقبلات الكيموكين لديهم أعلى احتمالات للبقاء على قيد الحياة على المدى الطويل أكثر من 90٪ مقابل أقل من 30٪ للمرضى الذين لديهم مستويات عالية جدا من المستقبلات.[20]
الانتشار
تمثل ساركومات يوينغ 16% من سرطانات العظام الأولية. في الولايات المتحدة،[21] تكون الأكثر شيوعًا في العقد الثاني من الحياة بمعدل 0.3 حالة لكل مليون في الأطفال دون سن 3 سنوات، وارتفاع يصل إلى 4.6 حالة لكل مليون في المراهقين الذين تتراوح أعمارهم بين 15-19 سنة. وعلى الصعيد الدولي، يبلغ معدل الإصابة السنوي أقل من حالتين لكل مليون طفل.[22] وفي المملكة المتحدة، يتم تشخيص ستة أطفال في المتوسط سنويًا ولا سيما الذكور في المراحل المبكرة من سن البلوغ.
كان أقدم مريض معروف شُخِّص في سن 76، من مقاطعة ميرسر في نيو جيرسي.[23]
تظهر ساركومة يوينغ اختلافات واضحة في حالات الإصابة بين البشر، وتتراوح بين 10 و20 ضعفًا أكثر شيوعًا بين السكان ذوي الأصل الأوروبي مقارنةً بالأفارقة.[24]
ساركومة يوينغ هو ثاني أكثر سرطان عظام شيوعًا في الأطفال والمراهقين.[25]
البحوث والمعلومات والدعم
في المملكة المتحدة وأيرلندا، تقوم مؤسسات أبحاث سرطان العظام ببحوث وتقدم معلومات عن ساركومة يوينغ وسرطانات العظام الأخرى. وهذا يشمل معلومات للمراهقين الذين لديهم هذه الحالة.[26]
مقالات ذات صلة
مراجع
- Iwamoto Y (February 2007). "Diagnosis and treatment of Ewing's sarcoma". Jpn. J. Clin. Oncol. 37 (2): 79–89. doi:10.1093/jjco/hyl142. PMID 17272319. مؤرشف من الأصل في 11 مايو 2020.
- Burt M, Karpeh M, Ukoha O, et al. (January 1993). "Medical tumours of the chest wall. Solitary plasmacytoma and Ewing's sarcoma". J. Thorac. Cardiovasc. Surg. 105 (1): 89–96. PMID 8419714. مؤرشف من الأصل في 14 أبريل 2013.
- Longtin R (November 2003). "Ewing sarcoma: a miracle drug waiting to happen?". J. Natl. Cancer Inst. 95 (21): 1574–6. doi:10.1093/jnci/95.21.1574. PMID 14600088. مؤرشف من الأصل في 14 أبريل 2020.
- synd/2367 على قاموس من سمى هذا؟
- Ewing, J. (1921). "Diffuse endothelioma of bone". Proceedings of the New York Pathological Society. 21: 17–24.
- "Symptoms of Ewing's Sarcoma - Bone Cancer Research Trust". Bcrt.org.uk. مؤرشف من الأصل في 30 يناير 201305 نوفمبر 2012.
- Owen LA, Kowalewski AA, Lessnick SL (2008). "EWS/FLI mediates transcriptional repression via NKX2.2 during oncogenic transformation in Ewing's sarcoma". PLoS ONE. 3 (4): e1965. doi:10.1371/journal.pone.0001965. PMC . PMID 18414662.
- Sorensen PH, Lessnick SL, Lopez-Terrada D, Liu XF, Triche TJ, Denny CT (February 1994). "A second Ewing's sarcoma translocation, t(21;22), fuses the EWS gene to another ETS-family transcription factor, ERG". Nat. Genet. 6 (2): 146–51. doi:10.1038/ng0294-146. PMID 8162068.
- Jeon IS, Davis JN, Braun BS, et al. (March 1995). "A variant Ewing's sarcoma translocation (7;22) fuses the EWS gene to the ETS gene ETV1". Oncogene. 10 (6): 1229–34. PMID 7700648.
- Bernstein M, Kovar H, Paulussen M, et al. (May 2006). "Ewing sarcoma family of tumours: current management". Oncologist. 11 (5): 503–19. doi:10.1634/theoncologist.11-5-503. PMID 16720851. مؤرشف من الأصل في 22 أغسطس 2009.
- "Soft tissue tumors: Ewing's tumors/Primitive neurectodermal tumors (PNET)". Atlas of Genetics and Cytogenetics in Oncology and Haematology. مؤرشف من الأصل في 29 أكتوبر 201205 نوفمبر 2012.
- Turc-Carel C, Aurias A, Mugneret F, et al. (June 1988). "Chromosomes in Ewing's sarcoma. I. An evaluation of 85 cases of remarkable consistency of t(11;22)(q24;q12)". Cancer Genet. Cytogenet. 32 (2): 229–38. doi:10.1016/0165-4608(88)90285-3. PMID 3163261.
- Delattre, Olivier; Zucman, Jessica; Plougastel, Béatrice; Desmaze, Chantal; Melot, Thomas; Peter, Martine; Kovar, Heinrich; Joubert, Isabelle; de Jong, Pieter (1992-09-10). "Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours". Nature. 359 (6391): 162–165. doi:10.1038/359162a0. PMID 1522903. مؤرشف من الأصل في 13 يوليو 2017.
- Lahl M, Fisher VL, Laschinger K (February 2008). "Ewing sarcoma family of tumours: an overview from diagnosis to survivorship". Clin J Oncol Nurs. 12 (1): 89–97. doi:10.1188/08.CJON.89-97. PMID 18258578. مؤرشف من الأصل في 28 يناير 2013.
- Randall, RL (2005). "Ewing's Sarcoma Family of Tumours (ESFT)". ESUN. مؤرشف من الأصل في 17 يونيو 201215 أبريل 2009.
- Abir R, Feinmesser M, Yaniv I, et al. (May 2010). "Occasional involvement of the ovary in Ewing sarcoma". Hum Reprod. 25 (7): 1708–12. doi:10.1093/humrep/deq121. PMID 20472912.
- McTiernan AM, Cassoni AM, Driver D, Michelagnoli MP, Kilby AM, Whelan JS (2006). "Improving Outcomes After Relapse in Ewing Sarcoma: Analysis of 114 Patients From a Single Institution". Sarcoma. 2006: 83548. doi:10.1155/SRCM/2006/83548. PMC . PMID 17496997.
- "ACS :: How Is the Ewing Family of Tumors Staged?". مؤرشف من الأصل في 22 أبريل 2008.
- Thacker, MM; Temple, HT; Scully, SP (2005). "Current treatment for Ewing's sarcoma". Expert review of anticancer therapy. 5 (2): 319–31. doi:10.1586/14737140.5.2.319. PMID 15877528.
- Bennani-Baiti IM; et al. (2010). "Intercohort gene expression co-analysis reveals chemokine receptors as prognostic indicators in Ewing's sarcoma". Clinical Cancer Research. 16 (14): 3769–3778. doi:10.1158/1078-0432.CCR-10-0558. PMC . PMID 20525755.
- medicine, s cecil. Goldman (الطبعة 24th). Philadelphia: Elsevier Saunders. صفحة 1326. .
- Ewing Sarcoma Imaging في موقع إي ميديسين
- WRAL (29 April 2013). "Three Wake students battle rare cancer: Cluster or coincidence? :: WRAL.com". مؤرشف من الأصل في 25 يونيو 2017.
- Worch, Jennifer; Cyrus, Jobin; Goldsby, Robert; Matthay, Katherine K.; Neuhaus, John; DuBois, Steven G. (2011-03-01). "Racial Differences in the Incidence of Mesenchymal Tumors Associated with EWSR1 Translocation". Cancer Epidemiology Biomarkers & Prevention. 20 (3): 449–453. doi:10.1158/1055-9965.EPI-10-1170. ISSN 1055-9965. PMC . PMID 21212061. مؤرشف من الأصل في 03 مايو 2019.
- Loeb, David M; Twardziok, Monika; Kleinsimon, Susann; Rolff, Jana; Jäger, Sebastian; Eggert, Angelika; Seifert, Georg; Delebinski, Catharina I. (2016). "Multiple Active Compounds from Viscum album L. Synergistically Converge to Promote Apoptosis in Ewing Sarcoma". PLOS ONE. 11 (9): e0159749. doi:10.1371/journal.pone.0159749. ISSN 1932-6203.
- Casado-Zapico, Sara, et al. "Synergistic Antitumor Effect Of Melatonin With Several Chemotherapeutic Drugs On Human Ewing Sarcoma Cancer Cells: Potentiation Of The Extrinsic Apoptotic Pathway." Journal of Pineal Research 48.1 (2010): 72-80. Academic Search Premier. Web. 5 Nov. 2016. نسخة محفوظة 20 مارس 2020 على موقع واي باك مشين.
وصلات خارجية
- Ewing family of tumors entry in the public domain NCI Dictionary of Cancer Terms