متلازمة كلاينفلتر (Klinefelter's syndrome) هي متلازمة توجد في الذكور الذين يمتلكون صبغ (X) (X chromosome) زائد في خلاياهم، ليصبح العدد 44+XXY بدلا من 44+XY. وسميت على اسم الدكتور هاري كلاينفلتر الذي وصفها لأول مرة عام 1942 [1]، وتحدث نتيجة عدم انتظام في توزيع الصبغيات أثناء عملية الانقسام الميوزي للبويضات في الأنثى حيث في بعض الحالات النادرة جدا يحدث ان تلتصق اصباغ (X) ببعضها أثناء الانقسام الميوزي بالتحديد عند الطور الاستوائي حيث بعدها لا تستطيع ازواج الكروموسومات ان تنفصل عن بعضها مما ينتج عنه بويضات تركيبها (22+xx)، وأخرى (22+0) فينتج عن اخصاب الأولى بحيوان منوي تركيبه (22+y) ذكر كلاينفلتر، اما ان كان تركيب الحيوان المنوي (22+x) فإن النتاج سيكون انثى عادية لديها حالة تضاعف جنسي.
| متلازمة كلاينفيلتر | |
|---|---|
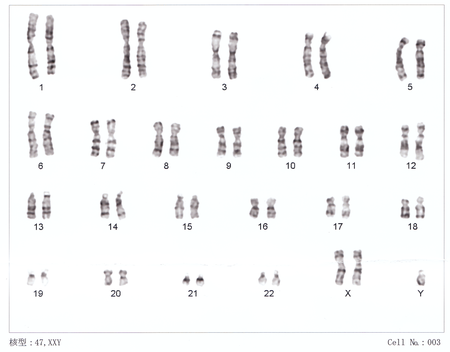 47,XXY
| |
| معلومات عامة | |
| الاختصاص | علم الوراثة الطبية |
| المظهر السريري | |
| الأعراض | فقد النطاف، وقصور الغدد التناسلية، وتثدي الرجل |
| الإدارة | |
| أدوية | |
| حالات مشابهة | متلازمة XYY |
| التاريخ | |
| سُمي باسم | هاري كلاينفلتر |

متلازمة كلاينفيلتر{47، XXY } أو متلازمة XXY هي الحالة التي يكون لدى الإنسان الذكر فيها كروموسوم الجنس X زائدا. فبينما يمتلك الإناث التكوين الكروموسومىXX والذكور XY يكون لدى الإنسان المصاب على الاقل اثنان من الكروموسوم X وكروموسوم Y واحد على الأقل. ونظرا لوجود هذا الكروموسوم الإضافي، فانه عادة ما يشار للأفراد المصابين بهذه الحالة باسم "XXYذكور "، أو "47، XXY ذكور".
ففي البشر، تعد متلازمة كلاينفيلتر أكثر اختلالات الصيغة الصبغية شيوعاً، وثاني أشهر الحالات الناجمة عن وجود الكروموسومات الزائدة. وهذه الحالة موجودة في حوالي 1 من كل 1000 من الذكور. واحد من500كروموسوم Xاضافي ولكن ليس لديه أعراض. وتوجد متلازمة XXYأيضا في ثدييات أخرى، بما في ذلك الفئران.
الآثار الرئيسية هي تكوين خصية صغيرة وانخفاض الخصوبة. وتوجد مجموعة متنوعة من الاختلافات الجسدية والسلوكية والمشكلات الشائعة، وعلى الرغم من تباين شدتها فان الكثير من الأولاد والرجال الذين يعانون من هذه الحالة لا يكتشف لديهم الا القليل من الاعراض.
ولقد تم تسمية المتلازمة باسم متلازمة الدكتور هاري كلاينفيلتر، الذي عمل في عام 1942 مع فولر أولبرايت في مستشفى ماساتشوستس العام في بوسطن، ماساشوستس، ووصفها لأول مرة في العام نفسه.[2]
معدل الحدوث
تحدث بنسبة 0.1% من المواليد الذكور فقط. وتعتبر من أشهر الاضطربات التي تصيب الصبغيات الجنسية[3].
العلامات والأعراض
ان الأعراض كلها عادة تكون نتيجة الصبغي (x) الزائد الذي أدى إلى اختلال في توازن هرمونات الجسم، وذلك حيث اننا نجد ان بعض الجينات الانثوية والمحمولة على ذات الصبغى قد عبرت بشكل ما عن نفسها وهذه الأعراض هي :
- العقم: وذلك نتيجة غياب الخلايا المولدة للحيوانات المنوية لديه.
- التأخر العقلي وصعوبة في التعلم والفهم.
- زيادة حجم الثدي، وعضلات الجسم تكون إلى حد ما انثوية.
- طول الأطراف أكثر من الطبيعي.
- شعر العانة يكون كشعر العانة للأنثى.
- اختفاء الصلع الأمامي (انحسار الشعر في جانبي الجبهة) الموجود لدى معظم الرجال.
- ضعف في الانتصاب وضعف الرغبة الجنسية.
- طول القامة.
- صغر الخصيتين[4].
تعانى الذكور المتضررة دائما تقريبا من العقم، على الرغم من إمكانية المساعدة الإنجابية المتقدمة في بعض الأحيان.[5] وقد توجد درجة من ضعف تعلم اللغة، [6] وتكشف الاختبارات النفسية والعصبية في كثير من الأحيان عن العجز في الوظائف التنفيذية.[7] في البالغين، تختلف الخصائص الممكنة على نطاق واسع وتتضمن القليل أو ما لا يذكر من علامات التأثر، ويكون الشخص المصاب نحيفاً وله بنية وملامح وجه شبابية، أو جسم من النوع المستدير مع درجة من التثدي (زيادة نسيج الثدي).[8] والتثدي يكون موجودا إلى حد ما في الثلث تقريبا من المصابين، وهي نسبة أعلى بقليل من الأشخاص الذين يمتلكون XX، ولكنه يكون ملاحظ في حوالي 10 ٪ فقط من 'الذكور XXY بحيث يتطلب عملية جراحية.
وفي كثير من الأحيان يساء فهم مصطلح hypogonadism في أعراض XXY على أنه يعني "الخصيتين الصغيرتين" أو "القضيب الصغير". وفي الواقع، فأنه يعني انخفاض هرمون الخصية / وظيفة الغدد الصماء. وبسبب هذا القصورالأولى في الغدد التناسلية فان الأفراد المصابين غالبا ما يكون لديهم مستوى منخفض من هرمون التستوستيرون في الدم بينما يكون لديهم مستوى مرتفع من هرمون تنشيط المسام (FSH)ولوتينيزينغ هورمون(LH) على الرغم من سوء فهم هذا المصطلح، فمن الصحيح أن الرجال XXY يكون لديهم خصيتين صغيرتين microorchidism
و تكون النهاية الأكثر شدة لهذا الطيف من الأعراض مرتبطة بزيادة خطر الإصابة بأورام الخلية الجرثومية وسرطان الثدي في الذكور وهشاشة العظام وتكون هذه المخاطر مشتركة بدرجات متفاوتة مع الإناث. بالإضافة إلى ذلك، تبين المؤلفات الطبية بعض دراسات الحالة الفردية لمتلازمة كلاينفيلتر مصاحبا لاضطرابات أخرى، مثل امراض الرئة، الدوالي، داء السكرى والتهاب المفاصل الروماتويدي ولكن الارتباطات المحتملة بين كلاينفيلتر وهذه الحالات الأخرى غير مفهومة،
وعلى الرغم من هذه المخاطر المتزايدة، فانه من المعتقد في الوقت الراهن أن الحالات المتنحية المرتبطة بالكروموسوم X تكون أقل شيوعا في الذكور من XXY عنها في الذكور الطبيعيين XY، حيث أن هذه الحالات تنتقل عن طريق الجينات المحمولة على الكروموسوم X فان الأشخاص الذين يمتلكون نسختان من الكروموسوم X يكونون فقط حاملين لا مصابين بهذه الحالات المتنحية المرتبطة بالكروموسوم X.
كما أن هناك فروقا عديدة بين أفراد XXY، تماما كما هو الحال في الأفراد 46,XY الأكثر شيوعا. في حين أنه من الممكن أن تميز الذكور XXY,47 بأنواع جسدية معينة، وهذا في حد ذاته لا يمكن أن يكون طريقة لتحديد ما إذا كان شخص ما 47، XXY أم لا. والطريقة الوحيدة الموثوقة لتحديد الهوية هي اختبار النمط النووي.
عدد الصبغيات
عدد الصبغيات في هذه الحالة قد يكون:-
- 47 صبغ ويكون هناك نسخة صبغ(X) واحدة زائدة فقط (44+xxy).
- 48 صبغ ويكون هناك نسختين من الصبغ (X)زائدة (44+xxxy).
- 49 صبغ ويكون هناك 3 نسخ من الصبغ (X) زائدة (44+xxxxy)، وهي حالات ناتجة عن تخصيب بويضات لانثي لديها تضاعف جنسي.
التشخيص
يتم استخدام النمط النووي للتأكد من التشخيص. في هذا الإجراء، تؤخذ عينة دم صغيرة. ثم يتم فصل خلايا الدم البيضاء من العينة، وخلطها مع وسط زراعة الأنسجة، ووضعها في الحضانة، والتحقق من شذوذ الكروموسومات، مثل الكروموسوم (X) الإضافي.
كما يمكن إجراء التشخيص قبل الولادة عن طريق أخذ العينات المشيمية أو البزل، الاختبارات التي يتم فيها استخراج أنسجة الجنين وفحص الحمض النووي لاكتشاف التشوهات الوراثية. وقد وجد مؤلفا عام 2002 عن معدلات الإجهاض الاختيارى أن ما يقرب من 50٪ من حالات الحمل في الولايات المتحدة مع التشخيص بمتلازمة Klinefelter قد أنهيت.
الأسباب
الاحتفاظ بكروموسوم Xاضافي بسبب حالة الانقسام المنصف {nondisjunction} خلال (انقسام خلية الجنس). الترتيب الكروموسومى XXY هو أحد أهم التغيرات الجينية الشائعة في النمط النووي XY، ويحدث في حوالي 1 لكل 500 من المواليد الذكور الأحياء.
في الثدييات التي تمتلك أكثر من كروموسوم X واحد فان الجينات التي تظهر هي تلك الموجودة على كروموسوم X واحد فقط دونا عن باقى الكروموسومات ؛و يعرف هذا بتعطيل الكروموسوم X. وهذا يحدث في الذكور XXY فضلا عن الإناث الطبيعيات XX. ومع ذلك، فان في الذكورXXY القليل من الجينات الموجودة في المنطقة الpseudoautosomal من الكروموسوم X له ما يقابله من الجينات على الكروموسوم Y وهي قادرة على الظهور. وهذه المضاعفة الثلاثية للجينات triploid في الذكور XXY قد تكون مسؤولة عن هذه الأعراض المصاحبة لمتلازمة Klinefelter.
ونشر التقرير الأول لرجل لديه النمط النووي XXY من قبل باتريشيا ألف جاكوبس، والدكتور ج. سترونج في المستشفى الغربي العام في ادنبره في اسكتلندا في عام 1959. وكان العثور على هذا النمط النووي في رجل يبلغ 24 عاما ولديه علامات متلازمة كلاينفيلتر. ووصفت الدكتور جاكوبس اكتشافها لعدم التوازن في الكروموسومات aneuploidy في الإنسان والثدييات لأول مرة أثناء خطابها في جائزة وليام ألان التذكارية عام 1981.
الاختلافات
و تحدث متلازمة 48, XXYY(الذكور)في 1 في كل 18,000-40,000 من المواليد وتقليديا تعتبر أحد أنواع كلاينفيلتر المختلفة. ولم يعد التضاعف الرباعى للكروموسومات XXYY tetrasomy يعتبر نوعا من أنواع متلازمة كلاينفيلتر بشكل عام بالرغم من عدم اعطائها الرمز 10 في التصنيف الدولي للأمراض حتى الآن.
و قد يكون لدى الذكور المصابين بمتلازمة كلايفيلتر النمط النووى الفسيفساءmosaic 47,XXY/46,XY مع درجات متفاوتة من عدم القدرة على تصنيع الحيوانات المنوية. ومن النادر جدا وجود حالات الفسيفساء Mosaicism 47,XXY/46,XX. بمظاهر سريرية توحي بوجود متلازمة كلايفيلتر. وحتى الآن، فقد وصفت حوالي 10 حالات فقط في المؤلفات الطبية.
العلاج
الاختلاف الجيني لا رجعة فيه. والتستوستيرون هو خيار العلاج لبعض الأفراد الذين يرغبون في مظهر وهوية أكثر ذكورة. وغالبا ما يعانى الأفراد الذين لديهم زيادة ملحوظة في نسيج الثدى أو انخفاض في هرمون الخصية hypogonadism من الاكتئاب أو/بالإضافة إلى القلق الاجتماعى وذلك لخروجهم عن المعايير الاجتماعية. يشار إلى هذا أكاديميا بالاعتلال النفسي والاجتماعى. وتشير دراسة واحدة على الأقل إلى أنه ينبغي توفير الدعم المخطط والمحدد زمنيا للشباب المصابين بمتلازمة كلاينفيلتر لتحسين النتائج النفسية والاجتماعية الضعيفة حاليا.
بحلول عام 2010 سيكون هناك ما يزيد عن 100 حمل ناجح باستخدام تقنية التلقيح الاصطناعى عن طريق المواد المنوية المزالة جراحيا من الرجال المصابين بمتلازمة كلاينفيلتر.
مقالات ذات صلة
المراجع
- Klinefelter, HF Jr; Reifenstein, EC Jr; Albright (1942), "Syndrome characterized by gynecomastia, aspermatogenesis without a-Leydigism and increased excretion of follicle-stimulating hormone", J Clin Endocrinol Metab, 2, صفحات 615–624 . Klinefelter, HF (1986), "Klinefelter's syndrome: historical background and development", South Med J, 79, صفحات 1089–1093, PMID 3529433 talks about the history of the development of the literature.
- Klinefelter, HF Jr; Reifenstein, EC Jr; Albright, F. (1942). "Syndrome characterized by gynecomastia, aspermatogenesis without a-Leydigism and increased excretion of follicle-stimulating hormone". J Clin Endocrinol Metab. 2: 615–624. doi:10.1210/jcem-2-11-615. . Klinefelter, HF (1986). "Klinefelter's syndrome: historical background and development". South Med J. 79 (45): 1089–1093. PMID 3529433. يتحدث عن تاريخ تطور الأدب.
- James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology. (10th ed.). Saunders. p 549. .
- Leask, Kathryn (2005). "Klinefelter syndrome". National Library for Health, Specialist Libraries, Clinical Genetics. National Library for Health. مؤرشف من الأصل في 27 سبتمبر 200707 أبريل 2007.
- Denschlag, Dominik, MD; Clemens, Tempfer, MD; Kunze, Myriam, MD; Wolff, Gerhard, MD; Keck, Christoph, MD (October 2004). "Assisted reproductive techniques in patients with Klinefelter syndrome: A critical review". Fertility and Sterility. 82 (4): 775–779. doi:10.1016/j.fertnstert.2003.09.085. PMID 15482743.
- Graham, JM Jr; Bashir, AS; Stark, RE; Silbert, A; Walzer, S (June 1988). "Oral and written language abilities of XXY boys: implications for anticipatory guidance". Pediatrics. 81 (6): 795–806. PMID 3368277.
- Boone KB, Swerdloff RS, Miller BL; et al. (2001). "Neuropsychological profiles of adults with Klinefelter syndrome". J Int Neuropsychol Soc. 7 (4): 446–56. doi:10.1017/S1355617701744013. PMID 11396547.
- [24] ^ خلاصة معلومات عن microorchidism (الخصيتين الصغيرتين). (العقم / العقم وظيفة هرمون الاندروجين) والتثدي. Bock, Robert (1993). "Understanding Klinefelter Syndrome: A Guide for XXY Males and Their Families". NIH Pub. No. 93-3202. Office of Research Reporting, NICHD. مؤرشف من الأصل في 29 يوليو 201828 مارس 2007. يقدم معلومات جوهرية عن نوع الجسم والمظهر حتى يتم العثور/الإمداد على مصدرأكثر صرامة.
قراءات أخرى
- Fales، سى ل، نولتون، ب. ج، Holyoak، كي ج، Geschwind، د ه، Swerdloff، آر س، وغونزالو، آى جى 2003). الذاكرة العاملة والعلاقات المنطقية في متلازمة كلاينفيلتر. مجلة الجمعية الدولية لعلم النفس العصبي، 9 ، 839-847.