وجود حمض الميثل مالونيك في الدم, هو اضطراب أيضي[1] جسمي متنحي[2] ويُعتبر نوع كلاسيكي من احمضاض الدم العضوي[3]. ينتج عن هذا الوضع عدم القدرة على هضم الدهون والبروتينات بشكل صحيح، مما يؤدي بدوره إلى تراكم مستوى سام من حمض الميثل مالونيك (بالانجليزية:methylmalonic acid) في الدم.[4]
| حمض الميثيل ملونيك | |
|---|---|
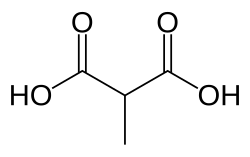 حمض الميثيل مالونيك
| |
| معلومات عامة | |
| الاختصاص | علم الغدد الصم |
| من أنواع | احمضاض الدم العضوي |
ينبعث مرض وجود حمض الميثل مالونيك في الدم من عدة أنماط جينية وراثية،[5] وجميع أشكال الاضطراب التي يتم تشخيصها في فترة حديثي الولادة عادة ما تُسبب اعتلال دماغي تدريجي، وفرط أمونيا الدم الثانوية. ناهيك عن أن هذا الاضطراب يمكن أن يؤدي إلى الوفاة إذا لم يتم تشخيصه أو إذا تم تركه دون علاج. وتشير التقديرات إلى أنه يحدث في واحد من كل 48000 ولادة، على الرغم من ان ارتفاع معدل الوفيات في الحالات التي تم تشخيصها يجعل التحديد الدقيق للاضطراب صعباً.[4] وقد تم العثور على هذا الاضطراب بتردد متساو عبر الحدود العرقية.[6]
أعراض المرض
حسب الجينات المتضررة، فان اعراض هذا الاضطراب تتراوح ما بين معتدلة إلى شديدة تهدد الحياة.
- السكتة الدماغية[4]
- اعتلال الدّماغ التّدريجي[4]
- نوبة مرضية (تشنّج)[4][7]
- الفشل الكلوي[8][4]
- القيء[4][7][8]
- الجفاف[4][7][8]
- الفشل في النماء والازدهار و التاخر في النمو [4][7][8]
- خمول [4][7][8]
- الاصابات الفطرية المتكررة [4]
- الحموضة[7]
- تضخم الكبد[7][8]
- نقص التوتر [7][8]
- التهاب البنكرياس[8]
- ضيق التنفس[7]
السبب
وراثي

ٍٍ

الأشكال الموروثة من وجود حمض الميثل مالونيك في الدم تسبب خللا في المسار الأيضي حيث يتم تحويل ميثل مالونيل-مرافق الانزيم أ إلى سكسينيل- مرافق الانزيم أ من خلال الانزيم ميثل مالونيل-مرافق الانزيم أ ميوتيز.[9]
هناك حاجة أيضا لوجود فيتامين بي 12 لتحويل ميثل مالونيل-مرافق الانزيم أ إلى سكسينيل- مرافق الانزيم أ. لذلك فان الطفرات التي تؤدي إلى خلل في التمثيل الغذائي لفيتامين بي 12 أو في نقله تكون سببا في تطوير وجود حمض الميثل مالونيك في الدم في كثير من الأحيان.
هذا الاضطراب الجسمي ناتج عن نمط طفرة متوارثة بشكل متنحي مما يعني ان الجين المعتل يكون محتوى داخل خلية جسمية , لذلك فان نسختين من الجين - واحد من كلا الوالدين - يجب أن تكون موروثة ليصاب الشخص بهذا الاضطراب. اما بالنسبة لوالدا الطفل الذي يعاني من وجود حمض الميثل مالونيك في الدم والذين يحملون نسخة واحدة من الجين المعيب، فغالبا ما يكونا غير مصابين بالاضطراب.
غذائي
على الرغم من أن السبب الغذائي ليس ذا صلة دائمة مع السبب الوراثي، الّا أن نقص التغذية الحاد من فيتامين بي 12 يمكن أن يؤدي أيضا إلى متلازمة مع أعراض وعلاجات متطابقة للاضرابات الجينية الناتج عن وجود حمض الميثل مالونيك في الدم.[10] ميثل مالونيل- مرافق الانزيم أ يحتاج فيتامين بي 12 لتشكيل سكسينيل- مرافق الانزيم أ. عندما تكون كمية فيتامين بي 12 غير كافية لتحويل العامل المساعد ميثل مالونيل- مرافق الانزيم أ إلى سكسينيل- مرافق الانزيم أ، فان تراكم ميثل مالونيل-مرافق الانزيم أ غير المستخدم يؤدي في نهاية المطاف إلى تراكم حمض الميثل مالونيك في الدم. وغالبا ما يستخدم هذا التشخيص كمؤشر على نقص فيتامين بي 12 في مصل الدم.[11]
الالية
فسيولوجيا المرض
عند الإصابة باضطراب وجود حمض الميثل مالونيك في الدم فان الجسم لا يستطيع تحطيم الاحماض الامينية (بالانجليزية: amino acids) مثل: المثيونين, ثريونين, ايزولوسين و فالين. كنتيجة لذلك يتراكم حمض الميثل مالونيك في الدّم و الانسجة.
المصابون بهذا المرض امّا يفتقرون إلى النّسخ الوظيفية أو يوجد لديهم عدد من الاحماض الامينية بشكل غير كافي مثل: ميثل مالونيل مرافق الانزيم أ ميوتيز (Methylmalonyl-CoA mutase) ميثل مالونيل مرافق الانزيم أ ايبماريز (بالانجليزية: methylmalonyl CoA epimerase) بالإضافة إلى تلك الانزيمات المسؤولة عن صناعة ال أدينوسيل كوبالامين(بالانجليزية: Adenosylcobalamin).[8][7]
ميثل مالونيل مرافق الانزيم أ ميوتيز
يقدّر انّ حوالي 60% من الحالات هي نتيجة طفرة حدثت في الجين الذي يترجم البروتين ميثل مالونيل مرافق الانزيم أ ميوتيز(بالانجليزية: Methylmalonyl-CoA mutase)،وهوالمسؤول عن هضم المشتقات السامة الناتجة من تكسر الاحماض الامينية والدهون، وبالمقام الأول ياتي الكولسترول.[8] بوجود الانزيم يتم تحويل ميثل مالونيل مرافق الانزيم أ إلى ساكسنال مرافق الانزيم أ (بالانجليزية: succinyl-CoA).[12] وعند نقص هذا الانزيم لا توجد اي وسيلة لتعديل تراكم الحمض أو ازالته أو المركبات الخاصّة فيه. وظيفة هذا الانزيم ايضا ممكن ان تتوقف نتيجة حدوث طفرات في كل من الجينات MMAA ,MMAB, و MMADHC , كلٌ منها مختص بترجمة بروتين معين بصورة طبيعية لتشغيل الميثل مالونيل مرافق الانزيم أ ميوتيز.[8]
ميثل مالونيل مرافق الانزيم أ ابيماريز
الطفرات في جين MCEE الذي يترجم البروتين ميثل مالونيل مرافق الانزيم أ ايبماريز، و يشار له ايضا بميثل مالونيل راسيميز يسبب ضرر اقل من الطفرة في جين الميوتيز. مع ان الايبماريز يعمل بنفس الطريقة عن طريق تكسير الاحماض الامينية و لكن بدرجة اقل .[8]
ويوجد ايضا بعض من الاختلافات في النمط الظاهري أو الصفات الجسمية نتيجة لنقص الايبيميريز، وهي معاكسة للميوتيز بحيث انها وصفت بالاعتدال في الاوساط الطبيّة، ويوجد نقاش عليها فيما ان يعتبر هذا النقص خللا وراثيا أو متلازمة سريرة ام لا.[13]
ادينوسيل كوبالامين
المعروف أيضا ب فيتامين بي 12, فان هذا الشكل من اشكال الكوبالمين مطلوب كعامل مساعد في ميثل مالونيل مرافق الانزيم أ ميوتيز. حتى بوجود كميات كافية و فعالة من الانزيم، إذا لم يتحول ال بي 12 إلى الصيغة الفعالة من، فان الميوتيز لن يكون قادر على العمل.[8]
تطوّر المرض
على الرّغم من عدم وجود مراحل متميزة للمرض، الا انّ هذا المرض متطور: اي انّ اعراض هذا الاضطراب تتضاعف نتيجة زيادة تركيز حمض الميثل مالونيك. وإذا لم يتم التخلص من البروتينات و الّدهون من الحمية الغذائية، فان هذا التراكم سيؤدي إلى الحاق الضّرر بالكلى أو الكبد وفي النّهاية الموت. [4]
التشخيص
اضطراب وجود حمض الميثل مالونيك في الدم، واحد من أشكال احمضاض الدم العضوي الأكثر شيوعا،[14] لا يظهر عند الولادة لأن الأعراض عادة لا تظهر من نفسها حتى تتم إضافة البروتينات إلى النظام الغذائي للرضع.[4] وبسبب هذا، تظهر الأعراض عادة في أي وقت خلال السنة الأولى من الحياة [14]. نظرا لشدة وسرعة هذا الاضطراب يمكن أن يسبب مضاعفات إذا تُرك من غير تشخيص، والكشف عن هذا الاضطراب غالبا ما يتم تضمينه في فحص لحديثي الولادة.[4][15]
بسبب عدم القدرة على تكسير الأحماض الأمينية تماما عند المصابين فانه يتم العثور على الناتج الثانوي من هضم البروتينات وحمض الميثل مالونيك المركب في تركيز غير متناسب في الدم والبول. وتستخدم هذه المستويات غير الطبيعية كمعايير التشخيص الرئيسية لتشخيص الاضطراب. وعادة ما يتم تحديد هذا الاضطراب من خلال استخدام تحليل البول أو الدم. [14] ويمكن أيضا أن يشتبه وجود حمض الميثل مالونيك في الدم من خلال استخدام التصوير المقطعي المحوسب أو التصوير بالرنين المغناطيسي (بالانجليزية: CT or MRI) أو اختبار الأمونيا، ولكن هذه الاختبارات ليست بأي حال من الأحوال مُحدِدة وتتطلب وجود الارتباط السريري والتمثيل الغذائي.[4] قد توجد مستويات مرتفعة من الأمونياك والجلايسين والاجسام الكيتونية أيضا في الدم والبول. [7]
الانواع
يتسم مرض وجود حمض الميثل مالونيك في الدم بتشخيصات متباينة ومتطلبات المعالجة والتنبؤ، والتي يتم تحديدها بواسطة طفرة جينية محدَدة والتي تسبب الشكل الوراثي للاضطراب.[5]
وفيما يلي الأنماط الجينية المعروفة المسؤولة عن هذا الاضطراب:
| OMIM | Name | Gene |
| 251100 | cblA type | MMAA |
| 251110 | cblB type | MMAB |
| 277400 | cblC type | MMACHC |
| 277410 | cblD type | [16]MMADHC |
| 277380 | cblF type | [17]LMBRD1 |
| 251000 | mut type | MUT |
نوع الطفرة (mut) يمكن أن يصنف ايضا إلى طفرة 0 وطفرات فرعية، مع تميز طفرة 0 بنقص كامل أو انعدام تام للانزيم لميثلمالونيل مرافق الانزيم أ ميوتيز وأعراض أكثر حدة امّا نوع الطفرات الفرعية فتتصف بانخفاض نشاط الانزيم ميوتيز.[6]
تم العثور على أنواع الطفرات Mut-, cblB, cblA من اضطراب وجود حمض الميثل مالونيك في الدم لتكون مستجيبة للكوبالامين على خلاف الطفرة 0 فانها متغير غير مستجيب.[6]
العلاج
النّظام الغذائي
يعتمد علاج جميع أشكال هذه الحالة في المقام الأول على نظام غذائي منخفض البروتينومكملات غذائية متنوعة على حسب نوع الاضطراب الذي يعاني منه الفرد. تستجيب جميع المتغيرات إلى ايزومرlevo isomer) levo )من كارنتين حيث يؤدي التفكك غير الملائم للمواد المتأثرة يؤدي إلى المعاناة من نقص في الكارنيتين في الأشخاص المصابة. وتساعد الكارنتين أيضًا في إزالة أسيل- مساعد الانزيم أ، والذي تراكمه شائع في النظام الغذائي منخفض البروتين، عن طريق تحويله إلى اسيل كارنيتين الذي يمكن إفرازه في البول. على الرغم من عدم استجابة جميع أشكال اضطراب وجود حمض الميثل مالونيك في الدم للكوبالامين، إلا أنسيانوكوبالامين تستخدم غالباً في المعالجة الأولى لهذا الاضطراب. إذا أثبت الشخص استجابةً لكل من مكمل الكوبالامين والكارنيتين[12]، فقد يكون من الممكن بالنسبة لهم تناول مواد تحتوي على كميات صغيرة من الأحماض الأمينية التي تسبب لهم مشاكل مثل السايولينيزين، والثريونين، والميثيونين، والفالين دون التسبب في هجوم.[4]
الجراحة
تشمل المعالجة الأكثر قصوى زرع الكلى أو الكبد من متبرع بدون شرط. تقوم الأعضاء المتبرع بها بإنتاج نسخة وظيفية من الإنزيمات المعيبة وهضم حمض الميثل مالونيك، ومع ذلك فإن كل عيوب زرع الأعضاء تنطبق بالطبع في هذه الحالة[4]. هناك أدلة تشير إلى أن الجهاز العصبي المركزي قد يستقلب ميثل مالونيك مرافق الانزيمأ في نظام معزول عن باقي الجسم. إذا كانت هذه هي الحالة، فإن زراعة الأعضاء قد لا تعكس التأثير العصبي لحمض الميثل مالونيك السابق لعملية الزرع أو تمنع حدوث المزيد من الضرر للدماغ عن طريق الاستمرار في البناء.[18][12]
التنبؤ بالمرض
يختلف التشخيص باختلاف شدة الحالة واستجابة الفرد للعلاج. عادة ما يكون التنبؤ أفضل بالنسبة لأولئك الذين يعانون من المتغيرات المستجيبة للكوبالامين وليست جيدة كثيرا في أولئك الذين يعانون من المتغيرات غير المستجيبة للكوبالامين [12]، وعادة ما تكون المتغيرات المعتدلة ذات تواتر أعلى في الظهور أكثر من المتغيرات الأكثر حدة.[14] حتى مع تعديل النظام الغذائي والرعاية الطبية المستمرة، قد لا يكون من الممكن منع حدوث ضرر عصبي في الأشخاص الذين يعانون من احمضاض غير مستجيب. [12] بدون علاج أو تشخيص مناسب، فانه من غير المألوف أن يكون الهجوم الحمضي الأول مميتًا.[4]
على الرغم من هذه التحديات، حيث تم تحديدها لأول مرة في عام 1967 ، فقد تحسن علاج وفهم الحالة إلى درجة كبيرة حتى لأولئك الذين يعانون من أشكال غير مستجيبة من اضطراب وجود حمض الميثل مالونيك في الدم حتى يتمكنوا من بلوغ سن الرشد وحتى حملهم وولادتهم للأطفال بأمان.[18]
أبحاث
تاريخ المرض
وجود حمض الميثل مالونيك في الدم تم وصفه اولا من قبل أوبيرهولزر ات ال[19] عام 1967 [18]
الاثار العصبية
يمكن أن يكون لوجود حمض الميثل مالونيك في الدم آثار كارثية على الجهاز العصبي؛ ومع ذلك، فإن الآلية التي يحدث فيها هذا لم تُحدد أبدا. أجريت البحوث على آثار حمض الميثل مالونيك على الخلايا العصبية وتم نشرها في 15 يونيو2015. كانت هذه الخلايا معزولة من جنين الفئران في إعداد في المختبر وتم مقارنته مع مجموعة من الخلايا العصبية التي وضع معها حمض بديل له درجة حموضة مماثلة. وقد اقترحت هذه الاختبارات أن حمض الميثل مالونيك يسبب انخفاض في حجم الخلايا وزيادة في معدل استماتة الخلايا بطريقة تعتمد على التركيز فنرى الآثار القصوى تظهر مع التراكيز الأعلى. وعلاوة على ذلك، اقترح تحليل مجموعة صغيرة من هذه الخلايا العصبية المعالجة أن حمض الميثل مالونيك على مستوى جيني, يغير معدل النسخ ل 564 جين، لا سيما تلك التي تشارك في موت الخلايا المبرمج، بي 53، ومسارات MAPK .[20]
الاختلال الوظيفي للميتوكندريا
بما أن تحول ميثل مالونيل-مرافق الانزيم أ إلى سكسينيل- مرافق الانزيم أ يحدث داخل الميتوكوندريات، فان اي خلل في وظيفة الميتوكوندريون نتيجة لانتقاص وظيفة سلسلة نقل الإلكترون يُشتبه به منذ فترة طويلة بان يكون ميزة لاضطراب وجود حمض الميثل مالونيك في الدم. أبحاث حديثة قد وجدت أن في نماذج الفئران، فان الميتوكوندريات من الفئران المتضررة من الاضطراب تنمو إلى حجم غير عادي، ويطلق عليها اسم ميغاميتوكوندريا. ويبدو أن هذه الميغاميتوكوندريا أيضا تتميز بتشوه الهياكل الداخلية وفقدان ثراء الإلكترون في مصفوفتها الداخلية. وأظهرت هذه الميغاميتوكوندريا أيضا علامات انخفاض وظيفة سلسلة الجهاز التنفسي، وخاصة في الجهاز التنفسي المجمع الرابع الذي يعمل فقط في حوالي 50٪ من الكفاءة. تم تحديد تغييرات مماثلة في الميتوكوندريات لعيّنة كبد تمت إزالتها خلال عملية زرع من صبي يبلغ من العمر 5 سنوات يعاني من وجود حمض الميثل مالونيك في الدم.[21]
نمط ظاهري حميد للطفرة
دراسات حديثة في العديد من المرضى الذين يعانون من نوع الطفرة 0 من اضطراب وجود حمض الميثل مالونيك في الدم مع طفرة محددة p.P86L قد تشير إلى إمكانية وجود مزيد من التقسيمات في نوع الطفرة من الاضطراب.على الرغم من عدم وضوح، في الوقت الراهن، ما إذا كان ذلك بسبب طفرة محددة أو الكشف المبكر والعلاج، وبالرغم من عدم الاستجابة الكاملة إلى مكملات كوبالامين، فيبدو أن هؤلاء الأفراد قاموا بتطوير نسخة حميدة وبدون أي اعراض ظاهرة لهذا الاضطراب. وبصرف النظر عن ظهور حمض الميثل مالونيك المرتفع في الدم والبول، إلا أن هؤلاء الأفراد ظهروا في الجزء الكبير من النمو الطبيعي.[22]
حالات ملحوظة
ريان ستالينغز، وهو طفل في سانت لويس، تم تشخيصه عن طريق الخطأ بتسمم الإيثيلين غليكول (بالانجليزية: Ethylene glycol poisoning) بدلا من وجود حمض الميثل مالونيك في الدم في عام 1989، مما أدى إلى إدانة القتل غير المشروع وحكم بالسجن مدى الحياة لأمه، باتريشيا ستالينغز (بالانجليزية: Patricia Stalling). [18]
المراجع
- "MMA Study: FAQ About Our Study". National Human Genome Research Institute (NHGRI) (باللغة الإنجليزية). مؤرشف من الأصل في 17 أكتوبر 201807 أبريل 2018.
- Radmanesh, Alireza; Zaman, Talieh; Ghanaati, Hossein; Molaei, Sanaz; Robertson, Richard L.; Zamani, Amir A. (2008-10-01). "Methylmalonic acidemia: brain imaging findings in 52 children and a review of the literature". Pediatric Radiology (باللغة الإنجليزية). 38 (10): 1054. doi:10.1007/s00247-008-0940-8. ISSN 0301-0449. مؤرشف من الأصل في 15 يونيو 2018.
- Dionisi-Vici, Carlo; Deodato, Federica; Röschinger, Wulf; Rhead, William; Wilcken, Bridget (2006-04-01). "Classical' organic acidurias, propionic aciduria, methylmalonic aciduria and isovaleric aciduria: Long-term outcome and effects of expanded newborn screening using tandem mass spectrometry". Journal of Inherited Metabolic Disease (باللغة الإنجليزية). 29 (2–3): 383–389. doi:10.1007/s10545-006-0278-z. ISSN 0141-8955. مؤرشف من الأصل في 15 يونيو 2018.
- "Methylmalonic acidemia: MedlinePlus Medical Encyclopedia". medlineplus.gov (باللغة الإنجليزية). مؤرشف من الأصل في 12 مايو 201907 أبريل 2018.
- Matsui, Suzanne M.; Mahoney, Maurice J.; Rosenberg, Leon E. (1983-04-14). "The Natural History of the Inherited Methylmalonic Acidemias". New England Journal of Medicine. 308 (15): 857–861. doi:10.1056/nejm198304143081501. ISSN 0028-4793. PMID 6132336. مؤرشف من الأصل في 16 ديسمبر 2019.
- "MMA Study: General Information". National Human Genome Research Institute (NHGRI) (باللغة الإنجليزية). مؤرشف من الأصل في 8 أبريل 201807 أبريل 2018.
- "Acidemia, Methylmalonic - NORD (National Organization for Rare Disorders)". NORD (National Organization for Rare Disorders) (باللغة الإنجليزية). مؤرشف من الأصل في 11 سبتمبر 201610 أبريل 2018.
- Reference, Genetics Home. "Methylmalonic acidemia". Genetics Home Reference (باللغة الإنجليزية). مؤرشف من الأصل في 9 مايو 201910 أبريل 2018.
- Sakamoto, Osamu; Ohura, Toshihiro; Matsubara, Yoichi; Takayanagi, Masaki; Tsuchiya, Shigeru (2007). "Mutation and haplotype analyses of the MUT gene in Japanese patients with methylmalonic acidemia". Journal of Human Genetics. 52 (1): 48–55. doi:10.1007/s10038-006-0077-2. ISSN 1434-5161. PMID 17075691. مؤرشف من الأصل في 11 أبريل 2018.
- Higginbottom, M. C.; Sweetman, L.; Nyhan, W. L. (1978-08-17). "A syndrome of methylmalonic aciduria, homocystinuria, megaloblastic anemia and neurologic abnormalities in a vitamin B12-deficient breast-fed infant of a strict vegetarian". The New England Journal of Medicine. 299 (7): 317–323. doi:10.1056/NEJM197808172990701. ISSN 0028-4793. PMID 683264. مؤرشف من الأصل في 19 أغسطس 2018.
- "Vitamins Problem Set". www.biology.arizona.edu. مؤرشف من الأصل في 4 مارس 201610 أبريل 2018.
- "Methylmalonic Acidemia: Brief Overview of Methylmalonic Acidemia, Etiology and Neuropathology, Evaluation of Methylmalonic Acidemia". 2018-03-13. مؤرشف من الأصل في 24 أبريل 2019.
- "OMIM Entry - # 251120 - METHYLMALONYL-CoA EPIMERASE DEFICIENCY". www.omim.org (باللغة الإنجليزية). مؤرشف من الأصل في 16 ديسمبر 201910 أبريل 2018.
- "Serials Solutions 360 Link". doi:10.4103/0972-5229.152776&rft.externaldbid=n/a&rft.externaldocid=10_4103_0972_5229_152776¶mdict=en-us. مؤرشف من الأصل في 16 ديسمبر 2019.
- "Newborn screening tests: MedlinePlus Medical Encyclopedia". medlineplus.gov (باللغة الإنجليزية). مؤرشف من الأصل في 24 مارس 201907 أبريل 2018.
- Coelho, David; Suormala, Terttu; Stucki, Martin; Lerner-Ellis, Jordan P.; Rosenblatt, David S.; Newbold, Robert F.; Baumgartner, Matthias R.; Fowler, Brian (2008-04-03). "Gene identification for the cblD defect of vitamin B12 metabolism". The New England Journal of Medicine. 358 (14): 1454–1464. doi:10.1056/NEJMoa072200. ISSN 1533-4406. PMID 18385497. مؤرشف من الأصل في 23 يوليو 2018.
- Rutsch, Frank; Gailus, Susann; Miousse, Isabelle R.; Suormala, Terttu; Sagné, Corinne; Toliat, Mohammad Reza; Nürnberg, Gudrun; Wittkampf, Tanja; Buers, Insa (February 2009). "Identification of a putative lysosomal cobalamin exporter altered in the cblF defect of vitamin B12 metabolism". Nature Genetics. 41 (2): 234–239. doi:10.1038/ng.294. ISSN 1546-1718. PMID 19136951. مؤرشف من الأصل في 11 أبريل 2018.
- "OMIM Entry - # 251000 - METHYLMALONIC ACIDURIA DUE TO METHYLMALONYL-CoA MUTASE DEFICIENCY". www.omim.org (باللغة الإنجليزية). مؤرشف من الأصل في 10 مايو 201710 أبريل 2018.
- Oberholzer, V. G.; Levin, B.; Burgess, E. A.; Young, W. F. (October 1967). "Methylmalonic aciduria. An inborn error of metabolism leading to chronic metabolic acidosis". Archives of Disease in Childhood. 42 (225): 492–504. ISSN 1468-2044. PMID 6061291. مؤرشف من الأصل في 11 أبريل 2018.
- "Serials Solutions 360 Link". ng4al8ll6x.search.serialssolutions.com. مؤرشف من الأصل في 10 مايو 201707 أبريل 2018.
- [Chandler, Randy j. (December 16, 2008). "Mitochondrial dysfunction in mut methylmalonic acidemia". The FASEB Journal. 23 (4): 1252–1261. doi:10.1096/fj.08-121848. Retrieved November 5, 2015 Chandler, Randy j. (December 16, 2008). "Mitochondrial dysfunction in mut methylmalonic acidemia". The FASEB Journal. 23 (4): 1252–1261. doi:10.1096/fj.08-121848. Retrieved November 5, 2015].
- "Serials Solutions 360 Link". ng4al8ll6x.search.serialssolutions.com. مؤرشف من الأصل في 10 مايو 201707 أبريل 2018.