
Les protéasomes sont des complexes enzymatiques multiprotéiques que l'on retrouve chez les eucaryotes, les archées ainsi que chez quelques bactéries de l'ordre Actinomycetales. Dans les cellules eucaryotes il se trouve dans le cytosol et est associé au réticulum endoplasmique[1] ainsi que dans le noyau. Leur fonction principale est de dégrader les protéines mal repliées, dénaturées ou obsolètes de manière ciblée. Cette dégradation se fait par protéolyse, une réaction chimique qui coupe les liaisons peptidiques et qui est effectuée par des enzymes appelées protéases. La protéine est ainsi découpée en peptides longs de 7 à 9 acides aminés qui seront ensuite hydrolysés hors du protéasome et recyclés[2]. Les protéines sont marquées pour la dégradation par une protéine appelée ubiquitine. Ce marquage est réalisé par l'action coordonnée de trois types d'enzymes. Une fois le marquage par une première molécule d'ubiquitine réalisé, d'autres ubiquitines vont être rajoutées à sa suite. Il faudra une chaîne d'au moins quatre ubiquitines pour que le protéasome 26S reconnaisse la protéine à dégrader. Il existe un compartiment pour celui-ci[2].
Le protéasome a une forme de baril et possède une cavité en son centre cernée par quatre anneaux, fournissant ainsi un espace clos pour la digestion des protéines. Chaque anneau est composé de sept protéines : les deux anneaux intérieurs sont constitués de sept sous-unités β qui contiennent le site actif de la protéase, tandis que les deux anneaux extérieurs contiennent sept sous-unités α dont le rôle consiste à maintenir l'ouverture par laquelle les protéines à dégrader pénètrent dans le baril: ces sous-unités α sont capables de reconnaître les marqueurs de polyubiquitine qui régulent le processus de dégradation. L'ensemble est connu sous le terme de complexe protéasome-ubiquitine.
La dégradation protéasomale est un élément essentiel de nombreux processus cellulaires, notamment le cycle cellulaire, l'expression génétique et la réponse au stress oxydatif. L'importance de la dégradation protéolytique et du rôle de l'ubiquitine lors de celle-ci a été officialisée par la remise du prix Nobel de chimie 2004 à Aaron Ciechanover, Avram Hershko et Irwin Rose[3]. La régulation du protéasome fait encore de nos jours l'objet de recherches intensives.
Découverte
Jusqu'à la découverte du complexe ubiquitine-protéasome, la dégradation des protéines dans les cellules était considérée comme essentiellement basée autour des lysosomes, des organites liés à la membrane cellulaire et dont l'intérieur acide est rempli de protéases qui peuvent dégrader et recycler les protéines exogènes et les organites usés ou endommagés[2]. De nouveaux travaux sur la dégradation protéique ATP-dépendante dans les réticulocytes, dépourvus de lysosomes, suggéraient la présence d'un mécanisme intracellulaire alternatif pour gérer cette dégradation: celui-ci, composé de plusieurs chaînes protéiques distinctes, fut décrit pour la première fois en 1978[4]. Des recherches subséquentes sur la modification des histones permit l'identification d'une modification covalente inattendue entre une lysine et une glycine N-terminale de l'ubiquitine, dont on ignorait à l'époque la fonction. On fit alors le lien avec une protéine récemment découverte et connue sous le nom de facteur protéolytique ATP-dépendant 1 (ATP-dependent proteolysis factor 1, ou plus simplement APF-1), qui ne faisait dès lors plus qu'un avec l'ubiquitine[5].

L'essentiel des premiers travaux qui menèrent à l'identification du protéasome eurent lieu dans les années 1970 et 80 au Technion, dans le laboratoire d'Avram Hershko où Aaron Ciechanover était doctorant. L'année sabbatique prise par Hershko au sein du groupe d'Irwin Rose au Fox Chase Cancer Center de Philadelphie permit de développer de nouvelles approches conceptuelles, bien que Rose ait largement relativisé son rôle dans cette découverte[6]. Quoi qu'il en soit, les trois chercheurs se sont partagé le Prix Nobel de chimie en 2004 pour leur découverte[3].
L'utilisation de la microscopie électronique au milieu des années 1980 permit de révéler la structure en anneaux empilés qui est celle du protéasome[7], mais la structure d'une des sous-unités (la 20S) ne fut résolue par cristallographie qu'en 1994[8]. Aucune structure complète montrant l'interaction entre centre actif et protéine cible n'a encore été résolue à ce jour (), même si des modèles ont été développés qui donnent une bonne idée du fonctionnement du complexe[9].
Structure et organisation
Les composants du protéasome sont nommés d'après leur coefficient de sédimentation de Svedberg (noté S). La forme la plus répandue est le protéasome 26S de masse moléculaire 2000 kilodaltons et qui contient un cœur catalytique en forme de baril, le 20S, et deux complexes régulateurs 19S placés de chaque côté de celui-ci.
Le centre est creux et forme la cavité au cœur de laquelle les protéines sont dégradées, après être entrées par l'une des deux extrémités, celle-ci s'associant avec une sous-unité régulatrice 19S contenant des sites actifs d'ATPase et des sites de liaison avec l'ubiquitine. Cette structure reconnaît les protéines polyubiquitinées et les transfère au centre catalytique. Une autre sous-unité (11S) peut également s'associer avec celui-ci, de la même manière que le ferait un complexe 19S : il a été postulé que 11S jouerait alors un rôle dans la dégradation de peptides « étrangers », tels que ceux produits lors de l'infection par un virus[10].
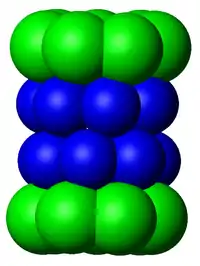
Cœur catalytique 20S
Le nombre et la variété des sous-unités constituant le complexe 20S dépend de l'organisme dont il fait partie, étant entendu que le nombre de sous-unités distinctes et leur degré de spécialisation est plus grand chez les multicellulaires que chez les unicellulaires, ainsi que chez les eucaryotes par rapport au procaryotes. Cependant, tous les 20S sont constitués de quatre structures heptamériques circulaires, elles-mêmes composées de deux types distincts de sous-unités: les deux anneaux extérieurs de sous unités α servent pour la régulation et ont un rôle plutôt structural (régulation de l'accès des protéines au cœur du protéasome), tandis que les sept sous-unités β des deux anneaux intérieurs portent l’activité catalytique par le biais des sites actifs des protéases qu'elles contiennent.
La taille du protéasome s'est relativement bien conservée pendant l'évolution, celui-ci mesurant environ 150 angstroms (Å) de long par 115 Å de diamètre. Le centre actif ne fait pas plus de 53 Å, mais il arrive que son chemin d'accès ne dépasse pas les 13 Å de large — il est donc probable que les protéines doivent être déjà partiellement dénaturées pour pouvoir interagir avec les sites actifs[11].
Chez les eucaryotes (comme la levure), les sept sous-unités sont toutes différentes les unes des autres, tandis que chez les archées elles sont toutes semblables. Les bactéries, à l'exception de l'ordre Actinomycetales, n’ont pas de protéasome 20S mais un complexe analogue appelé Hs1v avec un site catalytique ouvert. Chez les eucaryotes, seules trois sous-unités β sont actives mais elles ont des spécificités de coupure différentes : de type trypsine pour les acides aminés basiques, chymotrypsine pour les acides aminés aromatiques et caspase pour les acides aminés acides. Chez les mammifères par exemple, ce sont les sous-unités β1, β2, et β5 qui sont catalytiques[12]. Des variantes notées β1i, β2i et β5i peuvent être exprimées dans les cellules hematopoïétiques en réponse à des signaux inflammatoires tels que les cytokines, notamment les interférons gammas. Un protéasome constitué de ces sous-unités est appelé un immunoprotéasome, dont la spécificité de substrat est altérée par rapport à un protéasome « normal »[11].
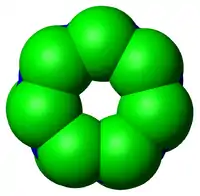
Complexe régulateur 19S
Le complexe 19S est constitué de 17 protéines distinctes et comporte deux zones : une « base » de 9 protéines se colle à l'anneau α du 20S, et possède l’activité ATPase sur 6 de ses 9 constituants (c'est la présence d'ATP qui permet l'association de 19S et 20S); l’autre partie (8 protéines), forme un « chapeau » plus flexible qui sert de couvercle à l’entrée dans le site catalytique. C’est ce complexe qui reconnaît la chaîne de polyubiquitine[13]. Il participe au dépliement de la protéine ainsi qu'à sa translocation dans le cœur du 20S, et contient également une isopeptidase qui va enlever la chaîne d’ubiquitine de la protéine, bien qu'on ne sache pas encore exactement si l'énergie dégagée par l'hydrolyse de l'ATP sert au dépliement[14], à l'ouverture du canal central[15] ou aux deux[13].
Les différents constituants du complexe 19S ont leur propre activité de régulation : La gankyrine, une oncoprotéine récemment découverte, est un élément de 19S qui interagit également avec la kinase cycline-dépendante CDK4, et joue un rôle dans la reconnaissance de la version ubiquitinée de p53 par le biais de son affinité avec l'ubiquitine ligase MDM2. La gankyrine est anti-apoptotique et il a été démontré qu'elle était surexprimée dans certains types de tumeurs tels que le carcinome hépatocellulaire[16].
Complexe régulateur 11S
Il existe enfin un complexe 11S heptamérique dit « activateur » qui peut s’associer au 20S. Il stimule l’activité peptidase du protéasome mais est incapable de reconnaître l’ubiquitine et ne possède pas d'activié ATPase. Il pourrait jouer un rôle dans la dégradation des peptides viraux, mais pas de protéines plus larges. Cette structure 11S peut parfois être rencontrée sous le terme de PA28 ou REG. Le mécanisme utilisé pour se lier à 20S par le biais de son C-terminus et ouvrir l'accès au cœur du protéasome en favorisant une modification de la conformation des anneaux α semble très proche, sinon similaire, à celui utilisé par le complexe 19S[17].
L'expression de 11S est induite par l'interféron gamma et, en association avec les sous-unités β de l'immunoprotéasome, provoque la génération de peptides qui iront se lier au complexe majeur d'histocompatibilité[10].
La structure du complexe 26S n'était toujours pas élucidée en 2006, mais on pense que les complexes 11S et 19S se lient de manière similaire au complexe 20S[15].
Assemblage
L'assemblage du protéasome est un processus complexe de par le nombre de sous-unités qui doivent s'associer afin de former un complexe actif. Les sous-unités β sont synthétisées avec un « propeptide » N-terminal qui sera modifié pendant l'assemblage du complexe 20S afin d'exposer le site actif protélytique. 20S est alors assemblé à partir de deux demi-protéasomes, chacun constitué d'un proto-anneau β à sept constituants et attaché à un heptamère α.
L'association des deux anneaux β déclenche une autolyse thréonine-dépendante des propeptides, exposant ainsi le site actif. Ces interactions entre β sont essentiellement le fait de ponts salins et d'interactions hydrophobes entre des hélices alpha conservées (leur mutation diminue la faculté d'assemblage du protéasome[18]). L'association des demi-protéasomes est initiée par le regroupement des sous-unités α en heptamère. Le mode d'assemblage de ces dernières n'a pas encore été caractérisé[19].
On connaît en général beaucoup moins le mode d'assemblage et de maturation du complexe régulateur 19S. Les sous-unités pourraient s'assembler en deux composants distincts, une base d'ATPases et un chapeau ubiquitine-spécifique. Les six ATPases s'assembleraient en paires par le biais d'interactions coiled coil[20]. L'ordre dans lequel les 19 sous-unités du complexe régulateur s'assemblent est probablement lié à un mécanisme empêchant l'exposition du site actif tant que le mécanisme de régulation n'est lui-même pas en place[15].
Processus de dégradation protéique

Ubiquitination
L’ubiquitine est une protéine de 76 acides aminés hautement conservée chez les Eucaryotes. Elle sert de signal de reconnaissance pour la dégradation par le protéasome 26S. Elle est fixée à la protéine à détruire par une liaison covalente entre l’un des résidus cystéine de sa partie C-terminale et un groupement NH2 d’une lysine de la protéine ciblée. D’autres ubiquitines peuvent alors se fixer à un résidu lysine de la première ubiquitine pour former une chaîne. En effet, seules les protéines liées à une chaîne d’au moins quatre molécules d’ubiquitine sont reconnues par le protéasome 26S.
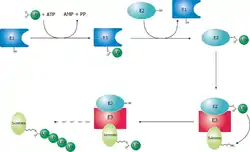
Ces opérations sont réalisés avec l’aide de trois autres types d’enzymes (notées E1,E2 et E3) :
- l’enzyme d’activation de l’ubiquitine (E1), active l’ubiquitine en présence d’ATP en formant une liaison thiolester entre un résidu cystéine de son site catalytique et l’ubiquitine, puis transfère l’ubiquitine activée sur l’une des E2s ;
- l’enzyme de conjugaison de l’ubiquitine(E2), modifie une fois de plus l’ubiquitine en formant une liaison thiolester et est capable d’attacher l’ubiquitine à la protéine cible en général avec l’aide d’une E3 ;
- l’ubiquitine ligase (E3), joue un rôle dans la reconnaissance entre l’ubiquitine et les différentes protéines cibles.
Un des domaines de l’ubiquitine reconnaît alors le complexe 19S du protéasome. Après avoir été enlevée par le protéasome, la chaîne de polyubiquitine est découpée pour être réutilisée. La monoubiquitination des protéines est également impliquée dans d’autres processus dont l’endocytose et l’exocytose.
Ubiquitination et ciblage
Les protéines sont ciblées pour être dégradées par le protéasome grâce à une modification covalente d'un résidu de lysine qui nécessite les actions coordonnées de trois enzymes. Dans un premier temps, une enzyme activatrice d'ubiquitine (connue sous le nom de E1) hydrolyse de l'ATP et adényle une molécule d'ubiquitine. Ensuite, la molécule est transférée vers les résidus de cystésine du site actif de E1, conjointement avec l'adénylation d'une seconde molécule d'ubiquitine. Cette molécule adénylée est par la suite transférée vers la cystéine d'une seconde enzyme (E2). Puis, un membre d'une classe d'enzymes très variées connues comme ligases d'ubiquitines (E3) reconnait la protéine spécifique qui doit être « ubiquinitée » et catalyse le transfert d'ubiquitine de E2 à la protéine ciblée. Une protéine cible doit être marquée avec au moins quatre monomères d'ubiquitine (sous la forme d'une chaine de polyubiquitine) avant d'être reconnue par une des deux extrémités du protéasome. C'est donc la protéine E3 qui confère le substrat spécifique au système. Le nombre des protéines E1, E2 et E3 exprimées dépend de l'organisme et du type de la cellule, mais il y a beaucoup d'enzymes E3 différentes présentes chez l'Homme, ce qui indique un nombre immense de cibles pour le système d'ubiquitine.
Le mécanisme par lequel une protéine polyubiquitinée est menée jusqu'au protéasome n'est pas pleinement compris. Des protéines réceptrices d'ubiquitine ont une zone de type « ubiquitine-like » (UBL) N-terminal et une ou plusieurs zones associées d'ubiquitine (UBA pour « ubiquitin-associated »). Les zones UBL sont reconnues par les capsules 19S du protéasome et les zones UBA lient l'ubiquitine via des paquets à trois hélices. Ces protéines réceptrices accompagnent probablement les protéines polyubiquitinées au protéasome, bien que les détails de ces interactions et leur régulation ne soient pas clairs.
La protéine d'ubiquitine est composée de 76 acides aminés et doit son nom à sa nature omniprésente, comme elle a une séquence hautement conservée et se trouve dans tous les organismes eucaryotes connus.
Les gènes codant l'ubiquitine dans les eucaryotes sont arrangés en paires, certainement à cause des importantes demandes de ces gènes de produire une quantité suffisante d'ubiquitine pour les cellules. L'hypothèse est émise que l'ubiquitine est la protéine la plus lente à évoluer identifiée à ce jour.
Déroulement et transfert
Texte anglais à traduire :
Ubiquitination and targeting
Proteins are targeted for degradation by the proteasome by covalent modification of a lysine residue that requires the coordinated reactions of three enzymes. In the first step, a ubiquitin-activating enzyme (known as E1) hydrolyzes ATP and adenylates a ubiquitin molecule. This is then transferred to E1's active-site cysteine residue in concert with the adenylation of a second ubiquitin[21]. This adenylated ubiquitin is then transferred to a cysteine of a second enzyme, ubiquitin-conjugating enzyme (E2). Lastly, a member of a highly-diverse class of enzymes known as ubiquitine ligases (E3) recognize the specific protein to be ubiquitinated and catalyze the transfer of ubiquitin from E2 to this target protein. A target protein must be labelled with at least four ubiquitin monomers (in the form of a polyubiquitin chain) before it is recognized by the proteasome lid[22]. It is therefore the E3 that confers substrate specificity to this system[23]. The number of E1, E2, and E3 proteins expressed depends on the organism and cell type, but there are many different E3 enzymes present in humans, indicating that there are a huge number of targets for the ubiquitin proteasome system.
The mechanism by which a polyubiquitinated protein is targeted to the proteasome is not fully understood. Ubiquitin-receptor proteins have an N-terminal ubiquitin-like (UBL) domain and one or more ubiquitin-associated (UBA) domains. The UBL domains are recognized by the 19S proteasome caps and the UBA domains bind ubiquitin via three-helix bundles. These receptor proteins may escort polyubiquitinated proteins to the proteasome, though the specifics of this interaction and its regulation are unclear[24].
The ubiquitin protein itself is 76 amino acids long and was named due to its ubiquitous nature, as it has a highly conserved sequence and is found in all known eukaryotic organisms. The genes encoding ubiquitin in eukaryotes are arranged in tandem repeats, possibly due to the heavy transcription demands on these genes to produce enough ubiquitin for the cell. It has been proposed that ubiquitin is the slowest-evolving protein identified to date[25].
Unfolding and translocation
After a protein has been ubiquitinated, it is recognized by the 19S regulatory particle in an ATP-dependent binding step[13]. The substrate protein must then enter the interior of the 20S particle to come in contact with the proteolytic active sites. Because the 20S particle's central channel is narrow and gated by the N-terminal tails of the α ring subunits, the substrates must be at least partially unfolded before they enter the core. The passage of the unfolded substrate into the core is called translocation and necessarily occurs after deubiquitination[13]. However, the order in which substrates are deubiquitinated and unfolded is not yet clear[26]. Which of these processes is the rate-limiting step in the overall proteolysis reaction depends on the specific substrate; for some proteins, the unfolding process is rate-limiting, while deubiquitination is the slowest step for other proteins[14]. The extent to which substrates must be unfolded before translocation is not known, but substantial tertiary structure, and in particular nonlocal interactions such as disulfide bonds, are sufficient to inhibit degradation[27].
The gate formed by the α subunits prevents peptides longer than about four residues from entering the interior of the 20S particle. The ATP molecules bound before the initial recognition step are hydrolyzed before translocation, although there is disagreement whether the energy is needed for substrate unfolding[14] or for gate opening[15]. The assembled 26S proteasome can degrade unfolded proteins in the presence of a non-hydrolyzable ATP analog, but cannot degrade folded proteins, indicating that energy from ATP hydrolysis is used for substrate unfolding[28]. Passage of the unfolded substrate through the opened gate occurs via facilitated diffusion if the 19S cap is in the ATP-bound state[29].
The mechanism for unfolding of globular proteins is necessarily general, but somewhat dependent on the amino acid sequence. Long sequences of alternating glycine and alanine have been shown to inhibit substrate unfolding decreasing the efficiency of proteasomal degradation; this results in the release of partially degraded byproducts, possibly due to the decoupling of the ATP hydrolysis and unfolding steps[30]. Such glycine-alanine repeats are also found in nature, for example in silk fibroin; in particular, certain Epstein-Barr virus gene products bearing this sequence can stall the proteasome, helping the virus propagate by preventing antigen presentation on the major histocompatibility complex[31].
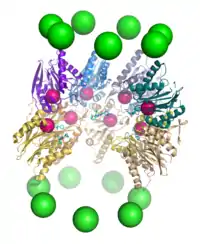
Proteolysis
The mechanism of proteolysis by the β subunits of the 20S core particle is through a threonine-dependent nucleophilic attack. This mechanism may depend on an associated water molecule for deprotonation of the reactive threonine hydroxyl. Degradation occurs within the central chamber formed by the association of the two β rings and normally does not release partially degraded products, instead reducing the substrate to short polypeptides typically 7–9 residues long, though they can range from 4 to 25 residues depending on the organism and substrate. The biochemical mechanism that determines product length is not fully characterized[32]. Although the three catalytic β subunits share a common mechanism, they have slightly different substrate specificities, which are considered chymotrypsine-like, trypsine-like, and peptidyl-glutamyl peptide-hydrolyzing (PHGH)-like. These variations in specificity are the result of interatomic contacts with local residues near the active sites of each subunit. Each catalytic β subunit also possesses a conserved lysine residue required for proteolysis[12].
Although the proteasome normally produces very short peptide fragments, in some cases these products are themselves biologically active and functional molecules. Certain transcription factors, including one component of the mammalian complex NF-κB, are synthesized as inactive precursors whose ubiquitination and subsequent proteasomal degradation converts them to an active form. Such activity requires the proteasome to cleave the substrate protein internally: rather than processively degrading it from one terminus. It has been suggested that long loops on these proteins' surfaces serve as the proteasomal substrates and enter the central cavity, while the majority of the protein remains outside[33]. Similar effects have been observed in yeast proteins; this mechanism of selective degradation is known as regulated ubiquitin/proteasome dependent processing (RUP)[34].
Ubiquitin-independent degradation
Although most proteasomal substrates must be ubiquitinated before being degraded, there are some exceptions to this general rule, especially when the proteasome plays a normal role in the post-translational processing of the protein. The proteasomal activation of NF-κB by processing p105 into p50 via internal proteolysis is one major example[33]. Some proteins that are hypothesized to be unstable due to intrinsically unstructured regions[35], are degraded in a ubiquitin-independent manner. The most well-known example of a ubiquitin-independent proteasome substrate is the enzyme ornithine decarboxylase[36]. Ubiquitin-independent mechanisms targeting key cell cycle regulators such as p53 have also been reported, although p53 is also subject to ubiquitin-dependent degradation[37]. Finally, structurally-abnormal, misfolded, or highly oxidized proteins are also subject to ubiquitin-independent and 19S-independent degradation under conditions of cellular stress[38].
Après qu'une protéine a été marquée par une chaîne d'ubiquitine, elle est reconnue par le complexe régulateur 19S qui s'y lie en consommant de l'ATP. Le substrat protéique doit ensuite entrer à l'intérieur du complexe 20S afin d'entrer en contact avec les sites actifs de l'enzyme. En raison de l'encombrement du pore central du complexe 20S dû aux extrémités N-terminales des sous-unités, le substrat doit être partiellement déplié afin de pouvoir pénétrer à l'intérieur du protéasome. Le passage de la protéine dépliée à l'intérieur du complexe 20S s'appelle la translocation et a lieu seulement après le retrait de la chaîne d'ubiquitine de la protéine. Il convient de préciser que l'ordre dans lequel la protéine est dépliée puis dé-ubiquitinée n'est pas encore bien clair.
Cible thérapeutique
Plusieurs molécules ont été développées comme médicaments agissant comme des inhibiteurs du protéasome. Le premier commercialisé est le bortézomib. Le protéasome est également une cible d'intérêt thérapeutique par le biais d'une nouvelle stratégie de développement de composés thérapeutiques. Il s'agit des molécules de types PROTAC qui vont, par le mécanisme d'action innovant, utiliser le protéasome pour induire une dégradation sélective d'une protéine cible[39],[40].
Notes et références
- ↑ Peters JM, Franke WW, Kleinschmidt JA. (1994) Distinct 19S and 20S subcomplexes of the 26S proteasome and their distribution in the nucleus and the cytoplasm. J Biol Chem, Mar 11;269(10):7709-18. .
- 1 2 3 Lodish, H, Berk A, Matsudaira P, Kaiser CA, Krieger M, Scott MP, Zipursky SL, Darnell J. (2004). Molecular Cell Biology, 5th ed., ch.3, p. 66-72. New York: WH Freeman. (ISBN 0716743663).
- 1 2 « Gagnants du Prix Nobel 2004, sur le site du Comité Nobel ».
- ↑ Ciehanover A, Hod Y, Hershko A. (1978). A heat-stable polypeptide component of an ATP-dependent proteolytic system from reticulocytes. Biochemical and Biophysical Research Communications 81(4):1100-1105. .
- ↑ Ciechanover A. (2000). Early work on the ubiquitin proteasome system, an interview with Aaron Ciechanover. Cell Death Differ. Sep;12(9):1167-77. .
- ↑ Hershko A. (2005). Early work on the ubiquitin proteasome system, an interview with Avram Hershko. Cell Death Differ. 12, 1158–1161. .
- ↑ Kopp F, Steiner R, Dahlmann B, Kuehn L, Reinauer H. (1986). Size and shape of the multicatalytic proteinase from rat skeletal muscle. Biochim Biophys Acta 872(3):253-60. .
- ↑ J. Löwe, D. Stock, B. Jap, P. Zwickl, W. Baumeister, R. Huber, « Crystal Structure of the 20S Proteasome from the Archaeon T. acidophilum at 3.4 Å Resolution », dans Science, vol. 268, 1995, p. 533-539. .
- ↑ Groll M, Huber R, Potts BC (2006). Crystal structures of Salinosporamide A (NPI-0052) and B (NPI-0047) in complex with the 20S proteasome reveal important conséquences of beta-lactone ring opening and a mechanism for irreversible binding. J Am Chem Soc. 128(15):5136-41. .
- 1 2 Wang J, Maldonado MA. (2006). The Ubiquitin-Proteasome System and Its Role in Inflammatory and Autoimmune Diseases. Cell Mol Immunol 3(4): 255. .
- 1 2 Nandi D, Tahiliani P, Kumar A, Chandu D. (2006). The ubiquitin-proteasome system. J Biosci Mar;31(1):137-55. .
- 1 2 Heinemeyer W, Fischer M, Krimmer T, Stachon U, Wolf DH. (1997). The Active Sites of the Eukaryotic 20S Proteasome and Their Involvement in Subunit Precursor Processing J Biol Chem 272(40):25200-25209. .
- 1 2 3 4 Liu CW, Li X, Thompson D, Wooding K, Chang TL, Tang Z, Yu H, Thomas PJ, DeMartino GN. (2006). ATP binding and ATP hydrolysis play distinct roles in the function of 26S proteasome. Mol Cell Oct 6;24(1):39-50. .
- 1 2 3 Lam YA, Lawson TG, Velayutham M, Zweier JL, Pickart CM. (2002). A proteasomal ATPase subunit recognizes the polyubiquitin degradation signal. Nature 416(6882):763-7. .
- 1 2 3 4 Sharon M, Taverner T, Ambroggio XI, Deshaies RJ, Robinson CV. (2006). Structural Organization of the 19S Proteasome Lid: Insights from MS of Intact Complexes. PLoS Biol Aug;4(8):e267. .
- ↑ Higashitsuji H, Liu Y, Mayer RJ, Fujita J. (2005). The oncoprotein gankyrin negatively regulates both p53 and RB by enhancing proteasomal degradation. Cell Cycle 4(10):1335-7. .
- ↑ Forster A, Masters EI, Whitby FG, Robinson H, Hill CP. (2005). The 1.9 Å Structure of a Proteasome-11S Activator Complex and Implications for Proteasome-PAN/PA700 Interactions. Mol Cell May 27;18(5):589-99. .
- ↑ Witt S, Kwon YD, Sharon M, Felderer K, Beuttler M, Robinson CV, Baumeister W, Jap BK. (2006). Proteasome assembly triggers a switch required for active-site maturation. Structure Jul;14(7):1179–88. .
- ↑ Kruger E, Kloetzel PM, Enenkel C. (2001). 20S proteasome biogenesis. Biochimie Mar-Apr;83(3-4):289-93. .
- ↑ Gorbea C, Taillandier D, Rechsteiner M. (1999). Assembly of the regulatory complex of the 26S proteasome. Mol Biol Rep Apr;26(1-2):15-9. .
- ↑ Haas AL, Warms JV, Hershko A, Rose IA. Ubiquitin-activating enzyme: Mechanism and role in protein-ubiquitin conjugation. J Biol Chem 1982 Mar 10;257(5):2543–8. .
- ↑ Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. (2000). Recognition of the polyubiquitin proteolytic signal. EMBO J 19, 94–102.
- ↑ Risseeuw EP, Daskalchuk TE, Banks TW, Liu E, Cotelesage J, Hellmann H, Estelle M, Somers DE, Crosby WL. Protein interaction analysis of SCF ubiquitin E3 ligase subunits from Arabidopsis. Plant J. 2003 Jun;34(6):753-67. .
- ↑ Elsasser S, Finley D. (2005). Delivery of ubiquitinated substrates to protein-unfolding machines. Nat Cell Biol 7(8):742-9.
- ↑ Sharp PM, Li WH. (1987). Ubiquitin genes as a paradigm of concerted evolution of tandem repeats. J Mol Evol 25(1):1432–1432.
- ↑ Zhu Q, Wani G, Wang QE, El-mahdy M, Snapka RM, Wani AA. (2005). Deubiquitination by proteasome is coordinated with substrate translocation for proteolysis in vivo. Exp Cell Res 307(2):436-51. .
- ↑ Wenzel T, Baumeister W. (1995). Conformational constraints in protein degradation by the 20S proteasome. Nat Struct Mol Biol 2: 199–204.
- ↑ Smith DM, Kafri G, Cheng Y, Ng D, Walz T, Goldberg AL. (2005). ATP binding to PAN or the 26S ATPases causes association with the 20S proteasome, gate opening, and translocation of unfolded proteins. Mol Cell 20(5):687-98.
- ↑ Smith DM, Benaroudj N, Goldberg A. (2006). Proteasomes and their associated ATPases: a destructive combination. J Struct Biol 156(1):72–83.
- ↑ Hoyt MA, Zich J, Takeuchi J, Zhang M, Govaerts C, Coffino P. (2006). Glycine-alanine repeats impair proper substrate unfolding by the proteasome. EMBO J 25(8):1720–9.
- ↑ Zhang M, Coffino P. (2004). Repeat sequence of Epstein-Barr virus-encoded nuclear antigen 1 protein interrupts proteasome substrate processing. J Biol Chem 279(10):8635–41.
- ↑ Voges D, Zwickl P, Baumeister W. (1999). The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem 68: 1015–1068.
- 1 2 Rape M, Jentsch S. (2002). Taking a bite: proteasomal protein processing. Nat Cell Biol 4(5):E113-6.
- ↑ Rape M, Jentsch S. (2004). Productive RUPture: activation of transcription factors by proteasomal processing. Biochim Biophys Acta 1695(1-3):209-13.
- ↑ Asher G, Reuven N, Shaul Y. (2006). 20S proteasomes and protein degradation "by default". Bioessays 28(8):844-9.
- ↑ Zhang M, Pickart CM, Coffino P. (2003). Determinants of proteasome recognition of ornithine decarboxylase, a ubiquitin-independent substrate. EMBO J 22(7):1488–96.
- ↑ Asher G, Shaul Y. (2005). p53 proteasomal degradation: poly-ubiquitination is not the whole story. Cell Cycle 4(8):1015–8.
- ↑ Shringarpure R, Grune T, Mehlhase J, Davies KJ. (2003). Ubiquitin conjugation is not required for the degradation of oxidized proteins by proteasome. J Biol Chem 278(1):311-8.
- ↑ Ian Churcher, « Protac-Induced Protein Degradation in Drug Discovery: Breaking the Rules or Just Making New Ones? », Journal of Medicinal Chemistry, vol. 61, no 2, , p. 444–452 (ISSN 1520-4804, PMID 29144739, DOI 10.1021/acs.jmedchem.7b01272, lire en ligne, consulté le )
- ↑ « De nouvelles molécules présentent un potentiel pour les futures thérapies contre le cancer », sur Clinique Amberieu : L'information médicale par des professionnels de la santé, (consulté le )
Voir aussi
- MG132
- Ubiquitine
- Ubiquitination