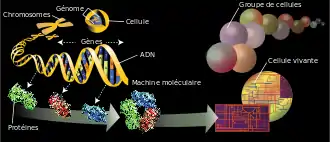
Le génome (/ʒenom/), ou plus rarement génôme, est l'ensemble du matériel génétique d'une espèce codé dans son acide désoxyribonucléique (ADN), à l'exception de certains virus dont le génome est constitué d'acide ribonucléique (ARN). Il contient en particulier tous les gènes codant des protéines ou correspondant à des ARN structurés. Il se décompose donc en séquences codantes (transcrites en ARN messagers et traduites en protéines) et non codantes (non transcrites, ou transcrites en ARN, mais non traduites).
Le génome est constitué de un ou plusieurs chromosomes dont le nombre total dépend de l'espèce considérée, chaque chromosome étant constitué d'une unique molécule d'ADN, linéaire chez les eucaryotes et le plus souvent circulaire chez les procaryotes. Chaque chromosome peut être présent en un ou plusieurs exemplaires, le plus souvent deux chez les espèces sexuées, l'un d'origine maternelle et l'autre d'origine paternelle (organisme diploïde).
La science qui étudie le génome est la génomique.
Il ne faut pas confondre le génome et le caryotype, qui est l'analyse ou la description macroscopique de l'arrangement des chromosomes.
Génomes dans le monde vivant

Chez les virus, le génome est contenu soit dans une ou plusieurs molécules d'ADN (virus à ADN) ou d'ARN (virus à ARN ou ribovirus), à simple ou double brin, protégé au sein d’une particule ou capside de nature protéique. Certaines personnes ne considèrent pas les virus comme des organismes mais comme des parasites moléculaires. En effet, ils se reproduisent en infectant des cellules vivantes dans lesquelles ils injectent leurs génome. Certains virus, comme les rétrovirus auxquels se rattache par exemple le VIH, ont un génome constitué d’ARN, qui se présente soit sous la forme d’un seul brin ou d’un double brin. Quel que soit l’acide nucléique, il est pour certains virus, sous une forme circulaire et pour d’autres, sous forme linéaire. Dans tous les cas, le génome viral existe au moins dans une des phases de son cycle cellulaire sous la forme d’un ADN double brin.
Chez les procaryotes (bactéries et archées), le génome est généralement contenu dans une molécule d'ADN circulaire. Peut aussi exister un génome extrachromosomique, contenu dans des plasmides et des épisomes. Certaines bactéries, comme les actinomycètes, ont cependant des génomes linéaires.
Chez les eucaryotes, on distingue :
- le génome nucléaire, contenu dans le noyau qui caractérise les eucaryotes. C'est de ce génome dont on parle en général quand on parle du génome d'un eucaryote (animal, plante, champignon, etc.) ;
- les génomes non nucléaires, contenus dans des organites :
- le génome mitochondrial, contenu dans les mitochondries chez la quasi-totalité des eucaryotes. Les mitochondries contiennent de multiples molécules d'ADNmt circulaires ressemblant à l'ADN des bactéries ;
- le génome chloroplastique, contenu dans les chloroplastes, chez les eucaryotes photosynthétiques (algues et plantes).
Chez quelques eucaryotes (par exemple la levure) sont aussi présents des plasmides (de taille réduite).
Chez l'homme en particulier (organisme eucaryote), le génome nucléaire est réparti sur 46 chromosomes, soit 22 paires d'autosomes et deux gonosomes (XX chez la femme, XY chez l'homme).
Taille du génome
La taille du génome se mesure en nombre de nucléotides, ou bases. La plupart du temps, on parle de pb (pour paire de bases, puisque la majorité des génomes est constituée de doubles brins d'ADN ou bien d'ARN). On emploie souvent les multiples kb (pour kilobase) ou Mb (mégabase), qui valent respectivement 1 000 et 1 000 000 bases. La taille du génome peut aussi être exprimée en pg (picogrammes), ce qui correspond à la masse d'ADN (haploïde) par cellule. 1 pg représente environ 1 000 Mb.
La taille du génome peut varier de quelques kilobases chez les virus à plusieurs centaines de milliers de Mb chez certains eucaryotes. La quantité d'ADN, contrairement à ce qui a été longtemps supposé, n'est pas proportionnelle à la complexité apparente d'un organisme. Les urodèles, les dipneustes, certaines fougères ou encore certains conifères comme les pins[1] ont des génomes plus de 10 fois plus grands que le génome humain. Ce constat est fréquemment appelé paradoxe de la valeur C.
Grands génomes
À ce jour, l'organisme vivant ayant le plus grand génome connu est la plante herbacée Paris japonica ; celui-ci est long d'environ 150 milliards de paires de bases, soit près de 50 fois la taille du génome humain[2].
Certaines amibes, comme Amoeba dubia pourraient avoir un génome encore plus grand, jusqu'à 200 fois plus grand que celui d'Homo sapiens. Cette détermination est toutefois contestée et pourrait être faussée par le fait que ces organismes unicellulaires phagocytent un grand nombre d'autres microorganismes dont elles ingèrent les chromosomes, ce qui vient contaminer la détermination de leur contenu exact en ADN[2].
Petits génomes
Le génome de Mycoplasma mycoides (en) ne comprend que 1,08 Mb dont 573 gènes. En 2010, une équipe de l'Institut J. Craig Venter synthétise ce génome et l'introduit dans une cellule de M. capricolum (en) privée de son matériel génétique, créant ainsi une nouvelle souche de M. mycoides dénommée JCVI-syn1.0[3]. En 2016, cette équipe réussit, en supprimant des gènes non essentiels, à réduire le génome de M. mycoides à 0,53 Mb et 473 gènes, créant ainsi une souche minimale dénommée JCVI-syn3.0, en fait une nouvelle espèce dénommée M. laboratorium. Ce génome est le plus petit de toutes les cellules connues capables de se reproduire. Sur les 473 gènes conservés, 149 ont une fonction inconnue[4].
Contenu des génomes
Les génomes sont constitués de régions codantes, qui correspondent aux gènes, et des régions non codantes. Les régions non codantes sont constituées des segments intergéniques et des introns à l'intérieur des gènes. Le séquençage de l'ADN permet d'établir l'enchaînement des nucléotides des brins d'ADN, afin de cartographier le génome.
Gènes
Le nombre des gènes dans le génome des organismes vivants varie beaucoup moins que la taille du génome. Chez la plupart des organismes vivants il est compris entre 1 000 et 40 000. Il n'est pas non plus corrélé à la complexité apparente des organismes. La paramécie, organisme cilié unicellulaire, possède ainsi un génome contenant plus de gènes que celui de l'homme[5]. Le tableau suivant donne la taille totale du génome (y compris les régions hétérochromatiques qui ne sont en général pas séquencées) et le nombre de gènes présents chez un certain nombre d'organismes dont le génome a été entièrement séquencé.
| Organisme | Nombre de gènes | Taille du génome |
|---|---|---|
| Haemophilus influenzae (bactérie) | 1 800[6] | 1,8 Mpb |
| Escherichia coli (bactérie) | 4 300[7] | 4,6 Mpb |
| levure de bière | 6 000[8] | 12,1 Mpb |
| Drosophile (insecte) | ~14 500[9] | 150,0 Mpb |
| Nématode | ~21 000 | 110,0 Mpb |
| Arabette (plante à fleur) | ~25 500 | 110,0 Mpb |
| Souris | ~22 000 | 2700,0 Mpb |
| Homme | ~22 000[10] | 3400,0 Mpb |
| Paramécie | ~40 000[5] | 72,0 Mpb |
Régions non codantes
Comme le nombre de gènes varie dans des proportions beaucoup plus limitées que la taille du génome, lorsque la taille du génome augmente (voir section précédente), la proportion du génome qui correspond aux régions codantes diminue. On observe une augmentation de la longueur des introns ainsi que des régions intergéniques. Les différents types de régions non codantes sont listés ci-dessous avec, à titre d'exemple, leur proportion dans le génome humain[11] qui est représentatif de la situation chez les mammifères :
- les introns dans les gènes. Dans le génome humain, les régions codantes (exons) représentent 1,5 % de la longueur totale du génome et les introns près de 26 % ;
- les pseudogènes qui représentent 1,5 % du génome humain ;
- les répétitions en tandem qui représentent 5 % du génome humain ;
- les répétitions dispersées qui représentent 45 % du génome humain ;
- l'hétérochromatine. Environ 10 % dans le génome humain ;
- les autres régions non codantes. Environ 11 % du génome humain.
En plus des gènes, les génomes contiennent en effet souvent des pseudogènes. Ce sont des séquences qui ont de nombreuses caractéristiques des gènes (séquences codantes, séquence promoteur, signaux d'épissage…), mais qui ne sont pas fonctionnelles et ne conduisent donc pas à la production d'une protéine. Ceci peut être la conséquence de mutations génétiques qui ont altéré sa séquence. Le génome humain contient ainsi environ 20 000 pseudogènes, soit pratiquement autant que de gènes fonctionnels. Souvent les pseudogènes sont des duplications d'un gène actif qui conserve la fonctionnalité pour la cellule. On dénombre ainsi plusieurs pseudogènes pour le cytochrome c dans notre génome, en plus du gène fonctionnel. Dans d'autres cas, la transformation d'un gène en pseudogène conduit à une perte de fonction, lorsque c'est la seule copie active qui est atteinte par des mutations. Dans notre génome, c'est le cas du gène codant la L-guluno-γ-lactone oxydase, une enzyme permettant la synthèse de l'acide ascorbique qui est devenu un pseudogène, ce qui fait que nous devons absorber de la vitamine C chaque jour dans notre alimentation, faute de pouvoir la synthétiser.
Dans les grands génomes, la plus grande partie des régions non codantes est constituée de séquences répétées et plus particulièrement de répétitions dispersées. Leur proportion augmente aussi avec la taille du génome. Dans le génome humain, ce taux est d'environ 45 %[10]. Il dépasse 80 % dans le génome du blé, qui est cinq fois plus grand que celui de l'homme.
Structure tridimensionnelle du génome
La configuration tridimensionnelle du génome a une importance fonctionnelle : l'enroulement (ou « condensation ») de l'ADN sur lui-même grâce aux histones permet de « ranger » une grande quantité d'information génétique dans le minuscule noyau d'une cellule, et il permet aussi à des parties éloignées de chromosomes de se toucher quand se forment des boucles d'ADN (ces boucles permettent à deux gènes éloignés d'agir de concert). Le chromosome peut être comparé à un collier de perles où chaque perle est un gène ou l'un des autres « morceaux » d'ADN, mais dont le fonctionnement ne serait pas « linéaire ». Dans ce cas, pour allumer ou éteindre un gène (une perle), ce gène doit être connecté avec l'ADN qui contrôle ou régule son activité ou qui doit agir de concert (une autre perle, d'une forme complémentaire). Cet autre gène peut être situé assez loin sur ce collier (ou même sur un collier voisin, c'est-à-dire un autre chromosome)[12].
Depuis des décennies, les biologistes moléculaires soupçonnaient fortement que la manière dont l'ADN se déroule et se condense tridimensionnellement dans le noyau joue un rôle-clé en permettant ces connexions, là où il faut et quand il faut, tout en décuplant les capacités d'interactions entre des gènes éloignés[12].
Depuis le début des années 2000 on comprend un peu mieux le lien entre les « astuces » biochimiques et topologiques et utilisées par le génome lors de ses changements de configuration, lors des différentes phases de la mitose et/ou de la méiose et dans son état condensé[12].
Des techniques biomoléculaires nouvelles sont en développement pour modéliser ou observer la position relative d'un seul morceau d'ADN (un gène par exemple) au regard d'autres gènes ou morceaux de l'ADN afin de définir un « interactome transcriptionnel » (qui serait une sorte de cartographie des relations fonctionnelles entre tous les gènes interagissant, de tous les chromosomes d'un même organismes) [12] ; et il faut encore ajouter à cette complexité celle de l'épigénétique ou des relations de transfert horizontaux de gènes d'une espèces à l'autre (chez les bactéries par exemple).
En 2009, Erez Lieberman Aiden[13], et ses collègues ont produit une méthode (modèle probabiliste) dite Hi-C[14] cherchant à représenter toutes les connexions simultanées ou possibles d'un génome. Ils se sont heurtés à un problème de résolution, faisant qu'ils ne pouvaient d'abord distinguer que deux compartiments, l'un renfermant de l'ADN actif et l'autre où les gènes tendaient à être éteints ; cette technique ne pouvait alors être utilisée que sur l'ADN déplié et retiré du noyau, ce qui conduisait à des résultats flous[12]. Ils ont donc cherché à cartographier les contacts entre gènes ou autres éléments du génome dans des noyaux intacts, via des méthodes apportant des informations bien plus de détaillées (passant d'une résolution de millions de bases à une résolution permettant d'observer des éléments de seulement 1000 bases (typique d'un gène). Des programmes informatiques sophistiqués ont alors pu produire des morceaux de « cartes 3D de l'ADN » (pour huit lignées de cellules humaines, dont cancéreuses ou de tissus de base, ainsi que pour une lignée de cellules cancéreuses de souris de laboratoire[15]).
Pour une lignée humaine de cellules de cancer lymphatique, par exemple, environ 4 900 000 000 contacts ont été détectés entre différents morceaux d'ADN ; pour d'autres types de cellules, le nombre de contacts a varié de 395 à 1 100 millions. Plus les contacts sont nombreux, plus les éléments en contact sont proches dans l'espace tridimensionnel[12].
En 2014, Rao, Huntley, Aiden, et leurs collègues concluent (dans la revue Cell[15]) que le génome est disposé en environ 10 000 boucles, avec dans chaque type de cellule une configuration différente correspondant à différents types de contacts entre fragments d'ADN. Ces différences de structure induisent différents patterns d'activité génique, définissant chaque type de cellule selon Aiden[12].
Au sein de cellules issues de donneuses (de sexe féminin), on a noté la formation de « boucles gigantesques dans l'un des chromosomes X ». Cette boucle pourrait avoir pour fonction de mettre en silence le second chromosome X afin de permettre le bon fonctionnement des gènes du chromosome X encore actif[12].
Le groupe a comparé les cartes 3D du génome de cellules cancéreuses de la souris et de cellules cancéreuses humaines. Ces cartes étaient très semblables, avec souvent de mêmes boucles, ce qui laisse penser que la structure tridimensionnelle qui définit un type spécifique de cellules n'a pas beaucoup changé chez les mammifères au cours de l'évolution[15].
La réalisation de cartes 3D complètes du génome de différentes espèces permettra aux chercheurs, médecins et à l'industrie biotechnologiques de mieux comprendre ou exploiter les génomes des espèces. Le laboratoire d'Aiden a déjà en 2014 créé une application et un portail dit « Juicebox »[16] avec un moteur de recherche fonctionnant à la manière de celui de Google Earth où des chercheurs peuvent localiser dans l'espace du génome un gène les intéressant et voir les contacts qu'il a avec la boucle d'ADN qu'il « touche ». Ces cartes devraient aussi pouvoir confirmer ou infirmer la fonction pressentie de certains gènes impliqués dans les maladies génétiques ou le fonctionnement normal de l'organisme.
Elles reposent aussi la question des effets directs ou indirects des gènes introduit — souvent au hasard — dans la topologie de l'ADN (par les moyens de la transgenèse).
Génomique
C'est la discipline scientifique qui étudie le fonctionnement d'un organisme, d'un organe, d'un cancer, etc. à l'échelle du génome et non d'un seul gène, avec :
- La génomique structurale (séquençage du génome entier) ;
- La génomique fonctionnelle (recherche de la fonction et de l'expression des gènes séquencés en caractérisant le transcriptome et le protéome.
Annotation des génomes
L’annotation d’un génome consiste à analyser la séquence nucléotidique qui constitue l’information brute pour en extraire l'information biologique. Cette analyse poursuit deux objectifs successifs, le premier est de localiser les gènes et les régions codantes et le second est, une fois ces gènes localisés, d'identifier ou de prédire leur fonction biologique. Ces deux étapes reposent initialement sur l'utilisation d'outils algorithmiques sophistiqués, dont le développement constitue l'un des champs de la bio-informatique.
Pour localiser les gènes, il existe différents outils complémentaires : des méthodes statistiques qui identifient les régions codantes sur la base de l'analyse de la fréquence des codons, des méthodes de recherche de motifs et en particulier les signatures caractéristiques du démarrage et de la fin, des jonctions entre les introns et les exons, séquences promotrices, terminatrices, sites de fixation du ribosome (RBS).
Pour prédire la fonction potentielle de ces gènes (leur attacher une étiquette, portant leur nom probable, leur fonction probable, leurs interactions probables), on utilise des programmes de recherche d'homologie de séquence. Lorsque le produit d'un gène prédit à des ressemblances avec une protéine connue, on en déduit en général une homologie probable de fonction[17]. On peut également identifier dans la séquence protéique prédite des motifs d'acides aminés caractéristiques de certaines classes de protéines (kinases, protéases…) ce qui peut permettre d'attribuer une fonction probable au gène correspondant. Ce type d'annotation est appelé annotation fonctionnelle.
L'annotation peut être automatique c'est-à-dire s’appuyer uniquement sur des algorithmes recherchant des similarités (de séquence, de structure, de motifs…), permettant de prédire (en fait deviner) la fonction d’un gène. Elle aboutit au transfert « automatique » de l’information figurant dans l’étiquette d’un gène « similaire » d’un génome déjà annoté au génome en cours d’annotation
L'annotation automatique initiale est parfois complétée par une annotation manuelle par des experts qui valident ou invalident la prédiction en fonction de leurs connaissances ou de résultats expérimentaux. Celle-ci peut ainsi éviter le transfert automatique d’erreurs et donc leur propagation, ce qui peut devenir le grand problème auquel devra se confronter la génomique, compte tenu de l'afflux massif de données issues en particulier, des nouvelles techniques de séquençage (voir pyroséquençage).
Actualité du séquençage
La revue Science et Vie du mois de [18] annonce que « Toutes les espèces devraient avoir leur génome séquencé en 2028 ». Le projet Earth BioGenome s'est donné cet objectif pour les espèces animales, végétales et d'autres espèces multicellulaires.
En 2023, l'Université Johns-Hopkins annonce avoir terminé le séquençage du chromosome Y, dernier des chromosomes humains dont le séquençage n'était pas achevé. Cette annonce conduit à considérer que le séquençage du génome humain est complet[19].
Notes et références
- ↑ (en) A.M. Morse, D.G.. Peterson, M.N. Islam-Faridi, K.E. Smith, Z. Magbuana, S.A. Garcia, T.L. Kubisiak, H.V. Anderson, J.E. Carlson, C.D. Nelson et J.M. Davis, « Evolution of genome size and complexity in Pinus », PLoS One, vol. 4, , e4332 (PMID 19194510, lire en ligne)
- 1 2 (en) J. Pellicer, M. Fay et I. J. Leitch,, « The largest eukaryotic genome of them all? », Botanical Journal of the Linnean Society, vol. 164, , p. 10–15 (lire en ligne).
- ↑ (en) Daniel G. Gibson, John I. Glass, Carole Lartigue, Vladimir N. Noskov, Ray-Yuan Chuang et al., « Creation of a Bacterial Cell Controlled by a Chemically Synthesized Genome », Science, vol. 329, no 5987, , p. 52-56 (DOI 10.1126/science.1190719, lire en ligne
 , consulté le ).
, consulté le ). - ↑ (en) Clyde A. Hutchison III, Ray-Yuan Chuang, Vladimir N. Noskov, Nacyra Assad-Garcia, Thomas J. Deerinck et al., « Design and synthesis of a minimal bacterial genome », Science, vol. 351, no 6280, (DOI 10.1126/science.aad6253, lire en ligne , consulté le ).
- 1 2 (en) J.M. Aury, O. Jaillon et L. Duret et al., « Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia. », Nature, vol. 444, , p. 171-178 (PMID 17086204)
- ↑ (en) R.D. Fleischmann et al., « Whole-genome random sequencing and assembly of Haemophilus influenza Rd », Science, vol. 269, , p. 496-512 (PMID 7542800)
- ↑ (en) FR Blattner, G Plunkett, CA Bloch, NT Perna, V Burland, M Riley, J Collado-Vides, JD Glasner, CK Rode, GF Mayhew, J Gregor, NW Davis, HA Kirkpatrick, MA Goeden, DJ Rose, B Mau et Y Shao, « The complete genome sequence of Escherichia coli K-12. », Science, vol. 277, , p. 1453-1462 (PMID 9278503)
- ↑ (en) A. Goffeau, B.G. Barrell, H. Bussey, R.W. Davis, B. Dujon, H. Feldmann, F. Galibert, J.D. Hoheisel, C. Jacq, M. Johnston, E.J. Louis, H.W. Mewes, Y. Murakami, P. Philippsen, H. Tettelin et S.G. Oliver, « Life with 6000 genes », Science, vol. 274, , p. 563-567 (PMID 8849441)
- ↑ (en) M.D. Adams et S.E. Celniker et al., « The genome sequence of Drosophila melanogaster », Science, vol. 287, , p. 2185-2195 (PMID 10731132)
- 1 2 (en) International Human Genome Sequencing Consortium, « Initial sequencing and analysis of the human genome », Nature, vol. 409, , p. 820-921 (PMID 11237011)
- ↑ (en) T.R. Gregory, « Synergy between sequence and size in large-scale genomics », Nat. Rev. Genet., vol. 6, no 9, , p. 699-708 (PMID 16151375, lire en ligne)
- 1 2 3 4 5 6 7 8 Elizabeth Pennisi (2014) News intitulée 3D map of DNA reveals hidden loops that allow genes to work together ; Revue Science, mise en ligne 11 décembre 2014
- ↑ Erez Lieberman Aiden] (sa page personnelle) est un biologiste travaillant maintenant au Baylor College of Medicine (BCM) de Houston (Texas)
- ↑ Yaffe, E., & Tanay, A. (2011). Probabilistic modeling of Hi-C contact maps eliminates systematic biases to characterize global chromosomal architecture. Nature genetics, 43(11), 1059-1065.
- 1 2 3 Suhas S.P. Rao, Miriam H. Huntley, Neva C. Durand, Elena K. Stamenova, Ivan D. Bochkov, James T. Robinson, Adrian L. Sanborn, Ido Machol, Arina D. Omer, Eric S. Lander, Erez Lieberman Aiden, (2014) A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping ; DOI:https://dx.doi.org/10.1016/j.cell.2014.11.021 (résumé)
- ↑ Portail 'Juicebox' (Visualization software for Hi-C data) ; AIden Lab / Center for Genome Architecture (Baylor College of Medicine & Rice University)
- ↑ F. Dardel et F. Képès, Bioinformatique : génomique et post-génomique, Editions de l'École Polytechnique, , 153-180 p. (ISBN 978-2-7302-0927-4, lire en ligne)
- ↑ Science-et-vie.com, « Science & Vie : premier magazine européen de l’actualité scientifique - Science & Vie », sur www.science-et-vie.com, (consulté le )
- ↑ « Le séquençage du génome humain est enfin complet », Sciences et Avenir, , p. 20
Annexes
Bibliographie
- Terence A. Brown, Génomes, Flammarion médecine-sciences, 2004.
- Génétique, gènes et génomes : Cours et questions de révision, ouvrage collectif par Jean-Luc Rossignol, Roland Berger, Jean Deutsch, Marc Fellous, Dunod, 2004.
- Stuart J. Edelstein, Des gènes aux génomes, Odile Jacob, 2002.
- Jean-Michel Petit, Sebastien Arico, Raymond Julien, « Le mini manuel de génétique », DUNOD, 4e édition, (ISBN 978-2-10-072753-7)
Articles connexes
- Chimiogénomique
- Duplication du génome
- Évolution moléculaire
- Programme génétique
- Projet Génome Humain
- Épigénome, épigénétique
- Genome Research
- Séquençage de l'ADN
Lien externe
- (en) Genomesize.com
Film/vidéographie
- Conférence intitulée « Génome et Cancer » par Mark Lathrop, pour l'Université de tous les savoirs (vidéo de 57 min), .
- A 3D Map of the Human Genome, sur Youtube, par « cellvideoabstracts », sur la base d'un article paru dans la revue Cells en