فرفرية هينوخ-شونلاين* هي مرضٌ شائٍع بين الأطفال، يُصيب الجلد والأغشية المخاطية وفي بعضِ الأحيان يُصيب أعضاءً أُخرى. يؤدي المرض إلى ظهور فرفريةٍ مجسوسةٍ في الجلد (والتي تكون عبارةً عن بقعٍ نزفية مُرتفعةٍ صغيرةٍ تحت الجلد)، كما يترافق أحيانًا مع ألمٍ في المفاصل والبطن. إذا تأثرت الكلى بالمرض، فَقَد يحدثُ فَقدٌ لكمياتٍ صغيرةٍ من الدم والبروتين في البول (أي بول دموي وبيلة بروتينية)، ولكن هذا الفَقد عادةً ما يَمرُ دون أن يُلاحَظ، أما في حالاتٍ أُخرى قليلة، فإنَّ تأثر الكلى يتحول إلى مرضٍ كلوي مزمن. غالبًا ما يُسبقُ هذا المرض بعدوى، مثل عدوى الحلق.
| فرفرية هينوخ-شونلاين | |
|---|---|
| Henoch–Schönlein purpura | |
 حالة شديدة من فرفرية هينوخ-شونلاين على قدم طفل وساقه وذراعه
| |
| تسميات أخرى | التهاب وعائي بالغلوبيولين المناعي أ،[1] فرفرية تأقية،[2] فرفرية روماتزمية،[2] فرفرية هينوخ-شونلين التأقانية |
| النطق | /ˈhɛnək ˈʃɜːnlaɪn,_ˈʃoʊnʔ/ |
| معلومات عامة | |
| الاختصاص | علم المناعة |
| من أنواع | التهاب وعائي بفرط التحسس، وفرفرية غير قليلة الصفيحات |
| المظهر السريري | |
| الأعراض | فرفرية، والتهاب المفاصل، وألم بطني |
| الإدارة | |
| المآل | جيد في معظم المرضى |
| حالات مشابهة | اعتلال الكلية بالغلوبيولين المناعي A |
| الوبائيات | |
| انتشار المرض | في الأطفال، 20 حالة لكل 100,000 طفل سنويًا |
| التاريخ | |
| سُمي باسم | يوهان لوكاس شونلاين، وإدوارد هاينريخ هينوخ |
فرفرية هينوخ-شونلاين هي التهابٌ وعائي شامل، يتميز بترسباتٍ من المركبات المناعية التي تحتوي الغلوبيولين المناعي أ (IgA)، وعلى الرغم من هذا إلا أنَّ السبب الدقيق لهذا المرض لا يزال مجهولًا. يختفي المرض عادةً عند الأطفال في غضونِ عدة أسابيع، ولا يتطلب أيَ علاجٍ باستثناء السيطرة على الأعراض، ولكن المرض قد يرجعُ مرةً أُخرى في ثلث الحالات، كما قد يُسبب تلفًا كلويًا غير قابلٍ للشفاء في حالةٍ واحدةٍ من كل 100 حالة. أما في البالغين فيختلف مآل المرض عن الأطفال. ومُتوسط مدة ظهور الآفات الجلدية في هذا المرض حوالي 27.9 شهرًا.[3] في مُعظم الحالات، فإنَّ هذا المرض يستمر في سلسلةٍ من الاختفاء والرجعة على فترةٍ طويلةٍ من الزمن، كما قد تظهر مُضاعفات أكثر في كل مرة.[4]
قام الطبيب الألماني إدوارد هاينريخ هينوخ ومعلمهُ يوهان لوكاس شونلاين بوصف المرض في عقد الستينات من القرن التاسع عشر، حيثُ سميَ المرض نسبةً لهما.[5] يُعتبر ويليام أوسلر أول من تعرف على الآلية التحسيسية لفرفرية هينوخ-شونلاين.[5]
التسمية
فُرْفُرِيَّةُ هِيْنُوخ-شُونْلاين^[6][7][8][9] (بالألمانية: Henoch–Schönlein purpura) وتُدعى اختصارًا HSP، وتسمى أيضًا:
- فرفرية هينوخ-شوينلاين[7] أو فرفرية هينوخ-شونلين[7] أو متلازمة هينوخ-شونلاين[7][8] أو فرفرية شونلاين-هينوخ[8] أو داء شونلاين-هينوخ.[8][2]
- التهاب وعائي بالغلوبيولين المناعي أ (IgA vasculitis)[1].
- فرفرية تَأَقِيَّة أو فرفرية تَأقانِيّة (Anaphylactoid purpura)[2].[10][11]
- فرفرية روماتزمية أو فرفرية رثوية (Purpura rheumatica)[2].[12]
- فرفرية هينوخ-شونلين التأقانية.[13]
الفُرْفُرِيَّةُ (Purpura) هو مصطلحٌ طبي يُقصد به ظهور بُقعٍ أرجوانية أو حمراء على الجلد، وهذه البقع لا تبيض عند الضغط عليها، وتحصل بسبب نزفٍ تحت الجلد.[14] سُميت نسبةً إلى اللون الأرجواني (purple) والذي يُسمى أيضًا بالفُرْفير.[15]
يُعرف قاموس حِتّي الطبي فرفرية هينوخ-شونلاين على أنها «فُرْفُرِيَّةُ أرجية، ضعيفةُ الاستِجابة للمُعالجة».[9] كما تُعرفها مكتبة لبنان ناشرون على أنها «فَرفَرية أرَجية مع أعراض مَفصِلية ضعيفة الاستجابة للمعالجة».[16]
الأعراض والعلامات
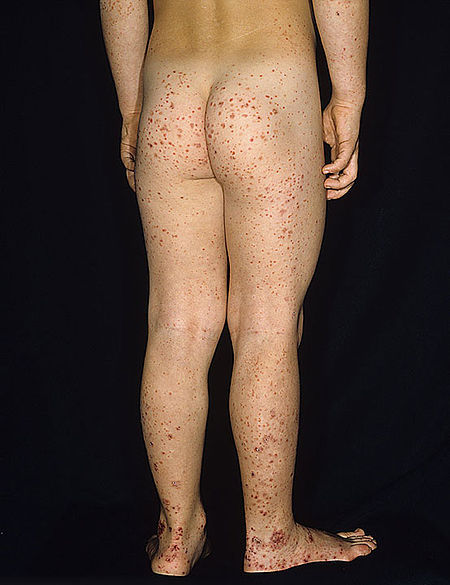
تتميز فرفرية هينوخ-شونلاين بثُلاثية من الأعراض المُترابطة وهي فرفرية والتهاب المفاصل وألم بطني.[17] تحدث الفرفرية في جميع الحالات، أما ألم والتهاب المفاصل في 80% والألم البطني في 62% من الحالات. يَعتبر البعض أنَّ النزف الهضمي هو العرض الرابع للمرض، حيثُ يحدث أحيانًا في 33% من الحالات، ولكن حدوثه ليس ضروريًا دائمًا، ويحدث بسبب انغلافٍ معوي.[18] تظهر الفرفرية عادةً على الساقين والأرداف، ولكنها قد تظهر أيضًا على الذراعين والوجه والجذع. يكون الألم البطني على شكل مغص، وقد يصاحبه غثيان أو قيء أو إمساك أو إسهال، كما قد يكون هناك دم أو مُخاط في البراز.[19] غالبًا ما تكون المفاصل المُتأثرة بالمرض هي الكاحل والركبة والمرفق، ولكن من المحتمل حدوث التهاب المفاصل في اليدين أو القدمين، فالتهاب المفاصل لا يؤدي للتآكل وبالتالي لا يسبب تشوهًا دائمًا.[17] قد تتأثر الكلية في 40% من الحالات، وغالبًا على شكل بول دموي، إلا أنَّ رُبع هذه الحالات فقط تكون فيها كمية الدم في البول كافية لملاحظتها دون إجراء فحوصاتٍ طبية.[18] قد يؤدي المرض لحدوث اضطراباتٍ في أعضاءٍ أُخرى، مثل الجهاز العصبي المركزي (الدماغ والحبل الشوكي) والرئتين، إلا أنَّ تأثر هذه الأعضاء أقل شيوعًا من تأثر الجلد والأمعاء والكلى.[20]
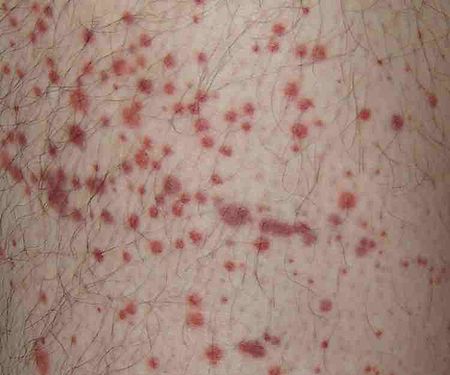
تقريبًا جميع المرضى الذين تتأثر الكلى لديهم (40% من الحالات)، يكون لديهم أدلة (مرئية أو تظهر في تحليل البول) على وجود الدم في البول، كما تحدث بيلة بروتينية في أكثر من نصف الحالات، وتكون هذه البيلة في ثُمن الحالات شديدةً كفاية لإحداث المتلازمة الكلوية (انتفاخٌ عام بسبب قلة البروتينات في الدم). على الرغم من أنَّ هذه الأدلة في تحليل البول قد تستمر لفترةٍ طويلة، إلا أنَّ 1% من الحالات فقط يحدث لديهم مرض الكلى المزمن.[20] قد يحدث ارتفاعٌ في ضغط الدم في بعض الحالات. يُساعد ارتفاع ضغط الدم وفقدان البروتين بالإضافة إلى إجراء خزعة للكلية، على توقع تطور المرض إلى مرضٍ كلويٍ مُتقدم. يكون البالغين أكثر عرضةً من الأطفال للإصابة بأمراض الكلى المتقدمة.[20][21]
الفيزيولوجيا المرضية
فرفرية هينوخ-شونلاين هي عبارةٌ عن التهابٍ وعائي صغير تترسب فيه مُركبات الغلوبيولين المناعي A (IgA) والمكون المتمم 3 (C3) على الشُرينات والوُريدات والشعيرات الدموية.[22] يُفترض أنَّ الاستعداد الوراثي والتعرض لمولدات الضد يؤدي إلى زيادة مستويات الغلوبيولين المناعي A في الدورة الدموية، مع تنشيط إنتاج الغلوبيولين المناعي G (IgG)، ثم تتفاعل هذه الغلوبيولينات مع بعضها البعض منتجةً مُركب يعمل على تنشيط المتممات، مع ترسبه في الأعضاء المُصابة، مسببًا رد فعلٍ التهابي مع التهابٍ وعائي.[23]
كما هو الحال في اعتلال الكلية بالغلوبيولين المناعي A، فإنَّ المستويات المصلية للغلوبيولين المناعي A تكون مُرتفعة في فرفرية هينوخ-شونلاين، وأيضًا توجد نتائج متماثلة في خزعة الكلية، ولكن اعتلال الكلية بالغلوبيولين المناعي A يميلُ للحدوث في الشباب البالغين أما فرفرية هينوخ-شونلاين تميل للحدوث في الأطفال. أيضًا اعتلال الكلية بالغلوبيولين المناعي A يؤثر عادةً على الكلى، أما فرفرية هينوخ-شونلاين فهي مرضُ جهازي شامل، حيثُ تتضمن الجلد والأنسجة الضامة وكيس الصفن والمفاصل والقناة الهضمية والكلى.[22]
التشخيص

يعتمدُ تشخيص فرفرية هينوخ-شونلاين على ظهور مجموعةٍ من الأعراض؛ وذلك لأنَّ عددًا قليلًا من الأمراض الأُخرى قد تسبب نفس الأعراض معًا. قد تُظهر تحاليل الدم ارتفاعًا في مستويات الكرياتينين واليوريا في حال تأثر الكلى، وقد يظهر ارتفاعٌ في مستويات الغلوبيولين المناعي A في حوالي 50% من الحالات،[22] كما قد ترتفعُ نتائج البروتين المتفاعل-C (CRP) وسرعة ترسب الدم (ESR)، ولكن جميع هذه التحاليل ليست خاصة بفرفرية هينوخ-شونلاين. قد يرتفع عدد الصفيحات الدموية أيضًا، وهذا يُساعد في تفريق فرفرية هينوخ-شونلاين عن غيرها من الأمراض التي يكون فيها انخفاضُ عدد الصفيحات الدموية هو المسؤول عن حدوث الفرفرية، مثل الفرفرية قليلة الصفيحات مجهولة السبب وفرفرية نقص الصفيحات التخثرية.
إذا كان هُناك شكٌ حول سبب حدوث الآفات الجلدية، فيُمكن إجراء خزعة من الجلد، بهدف تمييز الفرفرية عن غيرها من الأمراض التي قد تُسبب هذه الآفات مثل الالتهاب الوعائي بسبب وجود الغلوبيولينات البردية في الدم، وذلك لأنَهُ عند الاطلاع على الخزعة عبر المجهر يظهر التهاب الأوعية الصغيرة في الجلد، كما أنَّ التألق المناعي يحددُ وجود الغلوبيولين المناعي A والمكون المتمم 3 (بروتين ضمن جملة المتممة) في جدار الوعاء الدموي.[17] على الرغم من هذا، إلا أنَّ المستويات الإجمالية للجملة المتممة في مصل الدم تكون طبيعية.
يُمكن التمييز بين الالتهاب الوعائي بفرط التحسس (HV) وفرفرية هينوخ-شونلاين (HSP) اعتمادًا على الأعراض الظاهرة. قامت مجموعةٌ بمقارنة 85 مريضًا بفرفرية هينوخ-شونلاين مع 93 مريضًا بالالتهاب الوعائي بفرط التحسس، حيثُ وُجدَ أنَّ 5 أعراضٍ تُشير إلى الإصابة بفرفرية هينوخ-شونلاين وهي: فرفريةٌ مجسوسة، ذبحة بطنية، نزيف في القناة الهضمية (ليس بسبب انغلافٍ معوي)، بولٌ دموي، عمر أقل من 20 عامًا. وجودُ ثلاثةٍ أو أكثر من هذه الأعراض يُشير إلى حساسية بمقدار 87% لتوقع وجود فرفرية هينوخ-شونلاين.[24]
قد تُجرى خزعةٌ كلوية لتأكيد التشخيص أو لتقييم شدة المرض الكُلوي المُتوقع، وتتمثل نتائج هذه الخزعة في وجود زيادةٍ بترسيب الخلايا والغلوبيولين المناعي في مسراق الكبيبة (جُزءٌ من الكبيبة الكلوية، حيثُ يترشح فيه الدم)، كما تُوجد زيادة في خلايا الدم البيضاء مع ظهور الخلايا الهلالية. هذه النتائج لا يُمكن تمييزها عن تلك الموجودة في اعتلال الكلية بالغلوبيولين المناعي A.[22]

قد تحدثُ فرفرية هينوخ-شونلاين بعد العدواى بالعقدية (مجموعة لانسفيلد A، انحلالية بيتا)، التهاب الكبد ب، فيروس الهربس البسيط، الفيروس الصغير ب19، الفيروس الكوكساكي، الفيروسات الغدانية، الملوية البوابية،[20] الحصبة، النكاف، الحصبة الألمانية، المفطورة وغيرها.[22] مُعظم الأدوية المرتبطة بفرفرية هينوخ-شونلاين تمتلك رد فعلٍ ذاتي التحساس، وتتضمن المضادات الحيوية فانكوميسين وسيفيوروكزيم، ومثبطات الإنزيم المحول للأنجيوتنسين إنالابريل وكابتوبريل، والأدوية الديكلوفينية المضادة للالتهاب، بالإضافة إلى الرانيتيدين والستربتوكاينيز. وُثقت العديد من الأمراض المترافقة مع فرفرية هينوخ-شونلاين، وعادةً تحدث دون وجود رابطٍ مُسبب. في حوالي 35% فقط من الحالات يمكن أن يُعزى سبب حدوث هذه الفرفرية لأحد الأسباب السابقة.[22]
السبب الدقيق لحدوث فرفرية هينوخ-شونلاين غير معروفٍ حتى الآن، ولكن مُعظم ملامح هذا المرض تحدث بسبب ترسب الأجسام المضادة غير الطبيعية في جدار الأوعية الدموية، مما يؤدي لحدوث التهابٍ وعائي. تكون هذه الأجسام المضادة من النوع الفرعي الأول للغلوبيولين المناعي A (IgA1) في المبلمرات، ولكنه من غير المعروف فيما إذا كان السبب الرئيسي هو فرطُ إنتاج هذه الأجسام المضادة (في السبيل الهضمي أو نخاع العظم) أو انخفاض إزالة الغلوبيولين المناعي A من الدورة الدموية.[22] يُعتقد أنَّ التشوهات في جزيء IgA1 قد تُقدم تفسيرًا حول سلوكه الشاذ في فرفرية هينوخ-شونلاين وفي اعتلال الكلية بالغلوبيولين المناعي A. يتميز جزيء IgA1 (وIgD) باحتوائه على 18 حمض أميني تكون منطقة مفصلية طويلة بين المناطق 1 و2 المثبتة للمتممة. يكون نصف الأحماض الأمينية من البرولين أما النصف الآخر فيتكون بشكلٍ أساسي من السيرين والثريونين، حيثُ تحتوي معظم السيرينات والثريونينات على سلاسل سُكرية دقيقة، والتي ترتبط عبر ذرات الأكسجين (O-ارتباط بالغليكوزيل). يُعتقد أنَّ هذه العملية تعمل على استقرار جزيء IgA وتجعله أقل عرضةً للتحلل البروتيني. يكون السكر الأول دائمًا هو N-أسيتيل-جالاكتوزأمين (GalNAc)، ويليه الجالاكتوزات وحمض السياليك. في اعتلال الكلية بالغلوبيولين المناعي A وفرفرية هينوخ-شونلاين تكون سلاسل السكر هذه ناقصة، والسبب الدقيق لهذه التشوهات غير معروف.[20][22]
التصنيف
تُوجد معاييرٌ مُتعددة للتعرفِ على فرفرية هينوخ-شونلاين، وتتضمن تصنيف 1990 للكلية الأمريكية لأمراض الروماتيزم (ACR)،[25][26] وتصنيف 1997 لمؤتمر إجماع تشابل هيل (CHCC).[27] تُشير بعض التقارير أنَّ معايير الكلية الأمريكية لأمراض الروماتيزم أكثر حساسيةُ من معايير إجماع تشابل هيل.[28]
ظهرت تصنيفاتٌ حديثة للتعرفِ على فرفرية هينوخ-شونلاين، وتتضمن تصنيف 2006 للجمعية الأوروبية لمكافحة الروماتيزم (EULAR) وتصنيف جمعية أمراض الروماتيزم عند الأطفال (PReS)، حيثُ تعتبر أنَّ وجود الفرفرية المجسوسة معيارٌ إلزامي، مع وجود عرضٍ واحدٍ على الأقل من الأمور التالية: ألمٌ بطني منتشر، ترسبٌ دائم في الغلوبيولين المناعي A (يُؤكد عن طريق الخزعة)، التهابٌ مفصلي حاد في أيٍ مفصلٍ بالجسم، تضرر الكلية (حيثُ يظهر من خلال وجود دم و/أو بروتين في البول).[29]
التشخيص التفريقي
قد تحدثُ فرفرية هينوخ-شونلاين مع أعراضٍ غير مُعتادة (لانموذجية)، وهذا قد يؤدي إلى ارتباكٍ في تشخيصها أو خلطٍ مع أمراضٍ أُخرى مثل الشرى، والذئبة الحمامية الشاملة، ومرض المكورات السحائية، والتهاب الجلد الهربسي الشكل، والوذمة النزفية الحادة في الرُضع.[30]
العلاج
اعتبارًا من عام 2017، فإنَّ الطريقة المُثلى لعلاج فرفرية هينوخ-شونلاين تبقى محلًا للجدل.[31] يُمكن استعمال المسكنات مع آلام المفاصل والبطن، كما أنَّ العناية بالجرح ضرورية في حال حدوث موت بالجلد وتقرحات.[31] من غير المؤكد ما إذا كانت فرفرية هينوخ-شونلاين تحتاجُ إلى علاجٍ أكثر من مجرد التحكم في الأعراض، حيثُ أنَّ معظم الناس لا يتلقون علاجًا بسبب ارتفاع معدل الشفاء التلقائي. يختلف الخبراء فيما إذا كان يجب استعمال الكورتيكوستيرويدات كعلاجٍ روتيني مع فرفرية هينوخ-شونلاين،[31] ولكن على الرغم من هذا، إلا أنهُ إذا أُعطيت الكورتيكوستيرويدات مُبكرًا في نوبة المرض، فإنَّ مدة الأعراض قد تُصبح قصيرة ومن الممكن أن يتحسن ألم البطن بشكلٍ ملحوظ،[31] وأيضًا قد تقل فرصة حدوث مشاكل الكلى الشديدة.[32] أُجريت مراجعة منهجية حول استعمال الستيرويدات في العلاج، إلا أنها لم تجد أي دليلٍ على فعالية العلاج بالستيرويدات (بريدنيزون) في تقليل احتمال الإصابة بأمراض الكلى على المدى الطويل.[33]
عند وجودِ أي دليلٍ على تفاقم ضرر الكلى فإنهُ عادةً ما تُجرى خزعة كلوية، ويعتمد العلاج المُتبع على نتائج الخزعة، حيثُ يمكن استعمال علاجاتٍ متنوعة ما بين ستيرويدات تُعطى عبر الفم إلى مزيجٍ وريدي من الميثيلبريدنيزولون (ستيرويد) وسيكلوفوسفاميد وديبيريدامول ثُم يليها البريدنيزون، كما يُمكن استعمال مجموعاتٍ أُخرى تتضمن ستيرويدات/آزاثيوبرين، وستيرويدات/سيكلوفوسفاميد (مع أو بدون هيبارين ووارفارين). أحيانًا تستعمل المعالجة بالغلوبيولين المناعي (IVIG).[22]
لا يُوجد أي دليل جيد على أنَّ علاج الأطفال الذين يعانون من فرفرية هينوخ-شونلاين بالأدوية المضادة للصفيحات يمنع ضرر الكلى المستمر.[33] كما لا يوجد أيضًا أي دليلٍ على أن علاج الأطفال أو البالغين باستخدام السيكلوفوسفاميد يمنع الإصابة بأمراض الكلى الشديدة.[33] لا يوجد أي مُبرر لاستعمال العلاج بالهيبارين.[33]
مآل المرض
يكون المآل العام للمرض جيدًا في معظم المَرضى، حيثُ يُشفى معظمهم خلال 4 أسابيع من حدوث المرض،[34] كما أظهرت إحدى الدراسات أنَّ الشفاء من المرض يحدث في 94% من الأطفال و89% من البالغين، وقد يحتاج بعضهم إلى العلاج.[35] في الأطفال دون سن العاشرة، قد تتكرر الحالة في حوالي ثُلث الحالات، وعادةً ما يكون في غضون أربعةِ أشهر من الظهور الأولي للمرض.[18] تكرار حدوث المرض أكثر شُيوعًا عند الأطفال الأكبر سنًا والبالغين.[20]
تأثر الكلى
تتأثر الكلى في البالغين وتتطور الحالة إلى مرضٍ كلوي انتهائي (ESKD) أكثر من الأطفال. في سلسلةٍ من 37 مريضًا في المملكة المتحدة، فإنَّ 10 مرضى (أي 27%) تطورت الحالة لديهم إلى مرضٍ كلوي مُتقدم. يُعتبر حدوث البيلة البروتينية وارتفاع ضغط الدم، مع ظهور علاماتٍ مرضية مميزة (تغييرات هلالية، تليفات خلالية، ضمور أنبوبي) من النقاط المُهمة لتوقع تطور المرض.[21] حوالي 20% من الأطفال الذين تظهر لديهم علاماتٌ كلائية أو كلوية يعانون من ضعفٍ كلوي دائمٍ طويل الأمد.[36]
ترتبط نتائج الخزعة الكلوية مع شدةِ الأعراض: أولئك الذين يعانون من بيلةٍ دموية عديمة الأعراض قد يكون لديهم فقط تكاثرٌ مسراقي كبيبي بؤري بينما أولئك الذين لديهم بيلة بروتينية قد يكون لديهم تكاثرٌ خلوي أو حتى تكونٌ للهلاليات. يُعتبر عدد الكبيبات الهلالية عاملًا مهمًا في تشخيص ما إذا كان المريض سيصاب بمرضٍ كلوي مزمن.[20]
في حالِ حدوث مرضٍ كلوي انتهائي، فإنَّ البعض قد يحتاج إلى ديال دموي أو ما يُعادله من علاجٍ كلوي بديل (RRT). إذا أُجريت عملية زراعة الكلى للمريض كعلاجٍ بديل، فإنَّ نسبة حدوث المرض في الكلى المرزوعة تبلغ حوالي 35% من الحالات، وفي 11% من الحالات فإنَّ عملية الزراعة سوف تفشل تمامًا، ويجب الانتظار لإجراء زراعة أُخرى.[22]
الانتشار
حسب علم الوبائيات فإنَّ فرفرية هينوخ-شونلاين تحدثُ عادةً في الأطفال أكثر من البالغين،[35] في الفترة العمرية ما بين 3-10 سنوات،[23] ويعتبر سِن 5 سنوات العُمر المتوسط لظهور المرض.[34] عادةً ما يحدُث هذا المرض بعد حدوث عدوى بالجهاز التنفسي العلوي. نصف المرضى المصابين دون سن السادسة، و90% دون سن العاشرة. نسبة حدوث هذا المرض في الأولاد ضعف نسبته في الفتيات.[20] يبلغ معدل وقوع المرض في الأطفال حوالي 20 حالة لكل 100,000 طفل سنويًا، مما يجعله أكثر أنواع التهاب الأوعية الدموية شيوعًا في الأطفال.[23][37]
قد تحدث حالات فرفرية هينوخ-شونلاين في أي وقتٍ على مدار العام، ولكن وجدت بعض الدراسات أنَّ حالاتٍ أقل تحدث خلال أشهر الصيف.[38]
التاريخ
سُمي المرض نسبةً إلى إدوارد هاينريخ هينوخ (1820-1910)، وهو طبيبُ أطفالٍ ألماني ويكون ابن أخ موريتز هاينريخ رومبرغ، ومعلمهُ يوهان لوكاس شونلاين (1793–1864)، واللذان وصفا المرض في ستينات القرن التاسع عشر. قام شونلاين بالربط بين الفرفرية والتهاب المفاصل، أما هينوخ فربط بينها وبين التأثر الهضمي. كان الطبيب الباطني الإنجليزي وليام هيبيردين (1710–1801) قد وصف المرض عام 1802 وطبيب الجلد روبرت ويلان (1757–1812) قد وصفه عام 1808، وأُطلق عليه اسم مرض هيبيردين-ويلان (Heberden–Willan disease) ولكن هذه التسمية لم تعد مُستعملة الآن. يُعتبر ويليام أوسلر أول من تعرف على الآلية التحسسية لفرفرية هينوخ-شونلاين.[5]


مقالات ذات صلة
المراجع
- J. C. Jennette; R. J. Falk; P. A. Bacon; et al. (يناير 2013). "2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides". Arthritis & Rheumatism. 65 (1): 1–11. doi:10.1002/art.37715. PMID 23045170.
- Rapini RP, Bolognia JL, Jorizzo JL (2007). Dermatology. St. Louis: Mosby. .
- Sais G, Vidaller A, Jucglà A, Servitje O, Condom E, Peyri J (1998). "Prognostic factors in leukocytoclastic vasculitis: a clinicopathologic study of 160 patients". Dermatol. 134 (3): 309–15. PMID 9521029.
- "Treatment Challenges, Uncertainty Abound with IgA Vasculitis". The Rheumatologist. 2016. مؤرشف من الأصل في 16 ديسمبر 2018.
- Schönlein-Henoch purpura على قاموس من سمى هذا؟
- "ترجمة (Henoch–Schönlein purpura) في المعجم الطبي الموحد". المعجم الطبي الموحد. مؤرشف من الأصل في 22 مارس 201922 مارس 2019.
- "ترجمة (Henoch–schönlein purpura) على موقع القاموس". www.alqamoos.org. مؤرشف من الأصل في 17 ديسمبر 201922 مارس 2019.
- "ترجمة ومعنى henoch–schönlein purpura بالعربي في قاموس المعاني. قاموس عربي انجليزي مصطلحات صفحة 1". www.almaany.com. مؤرشف من الأصل في 22 مارس 201922 مارس 2019.
- يُوسف حِتّي; أحمَد شفيق الخَطيب (2008). قامُوس حِتّي الطِبي للجَيب. بيروت، لبنان: مكتبة لبنان ناشرون. صفحة 187. .
- "Al-Qamoos القاموس - English Arabic dictionary / قاموس إنجليزي عربي". www.alqamoos.org. مؤرشف من الأصل في 26 يناير 202022 مارس 2019.
- "ترجمة ومعنى anaphylactoid purpura بالعربي في قاموس المعاني. قاموس عربي انجليزي مصطلحات صفحة 1". www.almaany.com. مؤرشف من الأصل في 22 مارس 201922 مارس 2019.
- "Al-Qamoos القاموس - English Arabic dictionary / قاموس إنجليزي عربي". www.alqamoos.org. مؤرشف من الأصل في 17 ديسمبر 201922 مارس 2019.
- "ترجمة (Henoch–Schönlein purpura) في معجم مرعشي الطبي الكبير". معجم مرعشي الطبي الكبير. مؤرشف من الأصل في 22 مارس 201922 مارس 2019.
- "UCSF Purpura Module" ( كتاب إلكتروني PDF ). مؤرشف من الأصل ( كتاب إلكتروني PDF ) في 02 أكتوبر 2013.
- "LDLP - Librairie Du Liban Publishers". ldlp-dictionary.com. مؤرشف من الأصل في 22 مارس 201922 مارس 2019.
- "LDLP - Librairie Du Liban Publishers". ldlp-dictionary.com. مؤرشف من الأصل في 22 مارس 201922 مارس 2019.
- Kraft DM, Mckee D, Scott C (1998). "Henoch-Schönlein purpura: a review". American Family Physician. 58 (2): 405–8, 411. PMID 9713395. مؤرشف من الأصل في 6 يونيو 2011.
- Saulsbury FT (1999). "Henoch-Schönlein purpura in children. Report of 100 patients and review of the literature". Medicine (Baltimore). 78 (6): 395–409. doi:10.1097/00005792-199911000-00005. PMID 10575422.
- Fauci AS (1987). "269:The Vasculitis Syndromes". In Braunwald E, Isselbacher KJ, Petersdorf RG, Wilson JD, Martin JB, Fauci AS (المحررون). Harrison's Book of Internal Medicine. 2 (الطبعة 11th). McGraw Hill. صفحة 1441. .
- Saulsbury FT (2001). "Henoch-Schönlein purpura". Current Opinion in Rheumatology. 13 (1): 35–40. doi:10.1097/00002281-200101000-00006. PMID 11148713.
- Shrestha S, Sumingan N, Tan J, et al. (2006). "Henoch Schönlein purpura with nephritis in adults: adverse prognostic indicators in a UK population". QJM. 99 (4): 253–65. doi:10.1093/qjmed/hcl034. PMID 16565522. مؤرشف من الأصل في 29 يونيو 2009.
- Rai A, Nast C, Adler S (1 ديسمبر 1999). "Henoch-Schönlein purpura nephritis". Journal of the American Society of Nephrology. 10 (12): 2637–44. PMID 10589705. مؤرشف من الأصل في 3 يونيو 2009.
- Tom Lissauer; Will Carroll; Alan Craft (2018). Illustrated Textbook of Paediatrics (باللغة الإنجليزية) (الطبعة الخامسة). إلزيفير. صفحة 358-359. .
- Michel BA, Hunder GG, Bloch DA, Calabrese LH (1992). "Hypersensitivity vasculitis and Henoch-Schönlein purpura: a comparison between the 2 disorders". Journal of Rheumatology. 19 (5): 721–8. PMID 1613701.
- Mills JA, Michel BA, Bloch DA, et al. (1990). "The American College of Rheumatology 1990 criteria for the classification of Henoch-Schönlein purpura". Arthritis and Rheumatism. 33 (8): 1114–21. doi:10.1002/art.1780330809. PMID 2202310.
- American College of Rheumatology. "1990 criteria for the classification of Henoch-Schönlein purpura". مؤرشف من الأصل في 3 مارس 201615 ديسمبر 2007.
- Jennette JC, Falk RJ, Andrassy K, et al. (1994). "Nomenclature of systemic vasculitides. Proposal of an international consensus conference". Arthritis and Rheumatism. 37 (2): 187–92. doi:10.1002/art.1780370206. PMID 8129773.
- Murali NS, George R, John GT, et al. (2002). "Problems of classification of Henoch Schonlein purpura: an Indian perspective". Clinical and Experimental Dermatolology. 27 (4): 260–3. doi:10.1046/j.1365-2230.2002.01063.x. PMID 12139664.
- Ozen S, Ruperto N, Dillon MJ, et al. (يوليو 2006). "EULAR/PReS endorsed consensus criteria* for the classification of childhood vasculitides". Annals of the Rheumatic Diseases. 65 (7): 936–41. doi:10.1136/ard.2005.046300. PMC . PMID 16322081. مؤرشف من الأصل في 5 يوليو 2009.
- Lawee D (2008). "Atypical clinical course of Henoch-Schonlein purpura". Can Fam Physician (Review. Case Reports.). 54 (8): 1117–20. PMC . PMID 18697972.
- Hetland, LE; Susrud, KS; Lindahl, KH; Bygum, A (نوفمبر 2017). "Henoch-Schönlein Purpura: A Literature Review". Acta Dermato-venereologica (Review). 97 (10): 1160–66. doi:10.2340/00015555-2733. PMID 28654132.
- Weiss PF, Feinstein JA, Luan X, Burnham JM, Feudtner C (2007). "Effects of corticosteroid on Henoch-Schönlein purpura: a systematic review". Pediatrics. 120 (5): 1079–87. doi:10.1542/peds.2007-0667. PMC . PMID 17974746.
- Hahn, Deirdre; Hodson, Elisabeth M.; Willis, Narelle S.; Craig, Jonathan C. (2015-08-07). "Interventions for preventing and treating kidney disease in Henoch-Schönlein Purpura (HSP)". The Cochrane Database of Systematic Reviews (8): CD005128. doi:10.1002/14651858.CD005128.pub3. ISSN 1469-493X. PMID 26258874.
- Lloyd J. Brown; Lee Todd Miller (2005). Board Review Series Pediatrics (باللغة الإنجليزية). Lippincott Williams & Wilkins. صفحة 467-468. .
- Blanco R, Martínez-Taboada VM, Rodríguez-Valverde V, García-Fuentes M, González-Gay MA (1997). "Henoch-Schönlein purpura in adulthood and childhood: two different expressions of the same syndrome". Arthritis and Rheumatism. 40 (5): 859–64. doi:10.1002/art.1780400513. PMID 9153547.
- Watson, L; Richardson, AR; Holt, RC; Jones, CA; Beresford, MW (January 2012). "Henoch schonlein purpura--a 5-year review and proposed pathway". PLoS ONE. 7 (1): e29512. doi:10.1371/journal.pone.0029512. PMC . PMID 22235302.
- Gardner-Medwin JM, Dolezalova P, Cummins C, Southwood TR (2002). "Incidence of Henoch-Schönlein purpura, Kawasaki disease, and rare vasculitides in children of different ethnic origins". Lancet. 360 (9341): 1197–202. doi:10.1016/S0140-6736(02)11279-7. PMID 12401245.
- Saulsbury FT (2002). "Epidemiology of Henoch-Schönlein purpura". Cleveland Clinic Journal of Medicine. 69 Suppl 2: SII87–9. doi:10.3949/ccjm.69.suppl_2.sii87. PMID 12086273. مؤرشف من الأصل في 27 مارس 2020.